Chapter 9: Other Topics in Phase Equilibria
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Modelling and Numerical Simulation of Phase Separation in Polymer Modified Bitumen by Phase- Field Method
http://www.diva-portal.org Postprint This is the accepted version of a paper published in Materials & design. This paper has been peer- reviewed but does not include the final publisher proof-corrections or journal pagination. Citation for the original published paper (version of record): Zhu, J., Lu, X., Balieu, R., Kringos, N. (2016) Modelling and numerical simulation of phase separation in polymer modified bitumen by phase- field method. Materials & design, 107: 322-332 http://dx.doi.org/10.1016/j.matdes.2016.06.041 Access to the published version may require subscription. N.B. When citing this work, cite the original published paper. Permanent link to this version: http://urn.kb.se/resolve?urn=urn:nbn:se:kth:diva-188830 ACCEPTED MANUSCRIPT Modelling and numerical simulation of phase separation in polymer modified bitumen by phase-field method Jiqing Zhu a,*, Xiaohu Lu b, Romain Balieu a, Niki Kringos a a Department of Civil and Architectural Engineering, KTH Royal Institute of Technology, Brinellvägen 23, SE-100 44 Stockholm, Sweden b Nynas AB, SE-149 82 Nynäshamn, Sweden * Corresponding author. Email: [email protected] (J. Zhu) Abstract In this paper, a phase-field model with viscoelastic effects is developed for polymer modified bitumen (PMB) with the aim to describe and predict the PMB storage stability and phase separation behaviour. The viscoelastic effects due to dynamic asymmetry between bitumen and polymer are represented in the model by introducing a composition-dependent mobility coefficient. A double-well potential for PMB system is proposed on the basis of the Flory-Huggins free energy of mixing, with some simplifying assumptions made to take into account the complex chemical composition of bitumen. -

Phase Diagrams and Phase Separation
Phase Diagrams and Phase Separation Books MF Ashby and DA Jones, Engineering Materials Vol 2, Pergamon P Haasen, Physical Metallurgy, G Strobl, The Physics of Polymers, Springer Introduction Mixing two (or more) components together can lead to new properties: Metal alloys e.g. steel, bronze, brass…. Polymers e.g. rubber toughened systems. Can either get complete mixing on the atomic/molecular level, or phase separation. Phase Diagrams allow us to map out what happens under different conditions (specifically of concentration and temperature). Free Energy of Mixing Entropy of Mixing nA atoms of A nB atoms of B AM Donald 1 Phase Diagrams Total atoms N = nA + nB Then Smix = k ln W N! = k ln nA!nb! This can be rewritten in terms of concentrations of the two types of atoms: nA/N = cA nB/N = cB and using Stirling's approximation Smix = -Nk (cAln cA + cBln cB) / kN mix S AB0.5 This is a parabolic curve. There is always a positive entropy gain on mixing (note the logarithms are negative) – so that entropic considerations alone will lead to a homogeneous mixture. The infinite slope at cA=0 and 1 means that it is very hard to remove final few impurities from a mixture. AM Donald 2 Phase Diagrams This is the situation if no molecular interactions to lead to enthalpic contribution to the free energy (this corresponds to the athermal or ideal mixing case). Enthalpic Contribution Assume a coordination number Z. Within a mean field approximation there are 2 nAA bonds of A-A type = 1/2 NcAZcA = 1/2 NZcA nBB bonds of B-B type = 1/2 NcBZcB = 1/2 NZ(1- 2 cA) and nAB bonds of A-B type = NZcA(1-cA) where the factor 1/2 comes in to avoid double counting and cB = (1-cA). -

Phase Separation Phenomena in Solutions of Polysulfone in Mixtures of a Solvent and a Nonsolvent: Relationship with Membrane Formation*
Phase separation phenomena in solutions of polysulfone in mixtures of a solvent and a nonsolvent: relationship with membrane formation* J. G. Wijmans, J. Kant, M. H. V. Mulder and C. A. Smolders Department of Chemical Technology, Twente University of Technology, PO Box 277, 7500 AE Enschede, The Netherlands (Received 22 October 1984) The phase separation phenomena in ternary solutions of polysulfone (PSI) in mixtures of a solvent and a nonsolvent (N,N-dimethylacetamide (DMAc) and water, in most cases) are investigated. The liquid-liquid demixing gap is determined and it is shown that its location in the ternary phase diagram is mainly determined by the PSf-nonsolvent interaction parameter. The critical point in the PSf/DMAc/water system lies at a high polymer concentration of about 8~o by weight. Calorimetric measurements with very concentrated PSf/DMAc/water solutions (prepared through liquid-liquid demixing, polymer concentration of the polymer-rich phase up to 60%) showed no heat effects in the temperature range of -20°C to 50°C. It is suggested that gelation in PSf systems is completely amorphous. The results are incorporated into a discussion of the formation of polysulfone membranes. (Keywords: polysulfone; solutions; liquid-liquid demixing; crystallization; membrane structures; membrane formation) INTRODUCTION THEORY The field of membrane filtration covers a broad range of Membrane formation different separation techniques such as: hyperfiltration, In the phase inversion process a membrane is made by reverse osmosis, ultrafiltration, microfiltration, gas sepa- casting a polymer solution on a support and then bringing ration and pervaporation. Each process makes use of the solution to phase separation by means of solvent specific membranes which must be suited for the desired outflow and/or nonsolvent inflow. -

Interval Mathematics Applied to Critical Point Transitions
Revista de Matematica:´ Teor´ıa y Aplicaciones 2005 12(1 & 2) : 29–44 cimpa – ucr – ccss issn: 1409-2433 interval mathematics applied to critical point transitions Benito A. Stradi∗ Received/Recibido: 16 Feb 2004 Abstract The determination of critical points of mixtures is important for both practical and theoretical reasons in the modeling of phase behavior, especially at high pressure. The equations that describe the behavior of complex mixtures near critical points are highly nonlinear and with multiplicity of solutions to the critical point equations. Interval arithmetic can be used to reliably locate all the critical points of a given mixture. The method also verifies the nonexistence of a critical point if a mixture of a given composition does not have one. This study uses an interval Newton/Generalized Bisection algorithm that provides a mathematical and computational guarantee that all mixture critical points are located. The technique is illustrated using several ex- ample problems. These problems involve cubic equation of state models; however, the technique is general purpose and can be applied in connection with other nonlinear problems. Keywords: Critical Points, Interval Analysis, Computational Methods. Resumen La determinaci´onde puntos cr´ıticosde mezclas es importante tanto por razones pr´acticascomo te´oricasen el modelamiento del comportamiento de fases, especial- mente a presiones altas. Las ecuaciones que describen el comportamiento de mezclas complejas cerca del punto cr´ıticoson significativamente no lineales y con multipli- cidad de soluciones para las ecuaciones del punto cr´ıtico. Aritm´eticade intervalos puede ser usada para localizar con confianza todos los puntos cr´ıticosde una mezcla dada. -

Partition Coefficients in Mixed Surfactant Systems
Partition coefficients in mixed surfactant systems Application of multicomponent surfactant solutions in separation processes Vom Promotionsausschuss der Technischen Universität Hamburg-Harburg zur Erlangung des akademischen Grades Doktor-Ingenieur genehmigte Dissertation von Tanja Mehling aus Lohr am Main 2013 Gutachter 1. Gutachterin: Prof. Dr.-Ing. Irina Smirnova 2. Gutachterin: Prof. Dr. Gabriele Sadowski Prüfungsausschussvorsitzender Prof. Dr. Raimund Horn Tag der mündlichen Prüfung 20. Dezember 2013 ISBN 978-3-86247-433-2 URN urn:nbn:de:gbv:830-tubdok-12592 Danksagung Diese Arbeit entstand im Rahmen meiner Tätigkeit als wissenschaftliche Mitarbeiterin am Institut für Thermische Verfahrenstechnik an der TU Hamburg-Harburg. Diese Zeit wird mir immer in guter Erinnerung bleiben. Deshalb möchte ich ganz besonders Frau Professor Dr. Irina Smirnova für die unermüdliche Unterstützung danken. Vielen Dank für das entgegengebrachte Vertrauen, die stets offene Tür, die gute Atmosphäre und die angenehme Zusammenarbeit in Erlangen und in Hamburg. Frau Professor Dr. Gabriele Sadowski danke ich für das Interesse an der Arbeit und die Begutachtung der Dissertation, Herrn Professor Horn für die freundliche Übernahme des Prüfungsvorsitzes. Weiterhin geht mein Dank an das Nestlé Research Center, Lausanne, im Besonderen an Herrn Dr. Ulrich Bobe für die ausgezeichnete Zusammenarbeit und der Bereitstellung von LPC. Den Studenten, die im Rahmen ihrer Abschlussarbeit einen wertvollen Beitrag zu dieser Arbeit geleistet haben, möchte ich herzlichst danken. Für den außergewöhnlichen Einsatz und die angenehme Zusammenarbeit bedanke ich mich besonders bei Linda Kloß, Annette Zewuhn, Dierk Claus, Pierre Bräuer, Heike Mushardt, Zaineb Doggaz und Vanya Omaynikova. Für die freundliche Arbeitsatmosphäre, erfrischenden Kaffeepausen und hilfreichen Gespräche am Institut danke ich meinen Kollegen Carlos, Carsten, Christian, Mohammad, Krishan, Pavel, Raman, René und Sucre. -

Study of the LLE, VLE and VLLE of the Ternary System Water + 1-Butanol + Isoamyl Alcohol at 101.3 Kpa
View metadata, citation and similar papers at core.ac.uk brought to you by CORE Submitted to Journal of Chemical & Engineering Data provided by Repositorio Institucional de la Universidad de Alicante This document is confidential and is proprietary to the American Chemical Society and its authors. Do not copy or disclose without written permission. If you have received this item in error, notify the sender and delete all copies. Study of the LLE, VLE and VLLE of the ternary system water + 1-butanol + isoamyl alcohol at 101.3 kPa Journal: Journal of Chemical & Engineering Data Manuscript ID je-2018-00308r.R3 Manuscript Type: Article Date Submitted by the Author: 27-Aug-2018 Complete List of Authors: Saquete, María Dolores; Universitat d'Alacant, Chemical Engineering Font, Alicia; Universitat d'Alacant, Chemical Engineering Garcia-Cano, Jorge; Universitat d'Alacant, Chemical Engineering Blasco, Inmaculada; Universitat d'Alacant, Chemical Engineering ACS Paragon Plus Environment Page 1 of 21 Submitted to Journal of Chemical & Engineering Data 1 2 3 Study of the LLE, VLE and VLLE of the ternary system water + 4 1-butanol + isoamyl alcohol at 101.3 kPa 5 6 María Dolores Saquete, Alicia Font, Jorge García-Cano* and Inmaculada Blasco. 7 8 University of Alicante, P.O. Box 99, E-03080 Alicante, Spain 9 10 Abstract 11 12 In this work it has been determined experimentally the liquidliquid equilibrium of the 13 water + 1butanol + isoamyl alcohol system at 303.15K and 313.15K. The UNIQUAC, 14 NRTL and UNIFAC models have been employed to correlate and predict LLE and 15 16 compare them with the experimental data. -

Definitions of Terms Relating to Phase Transitions of the Solid State
Definitions of terms relating to phase transitions of the solid state Citation for published version (APA): Clark, J. B., Hastie, J. W., Kihlborg, L. H. E., Metselaar, R., & Thackeray, M. M. (1994). Definitions of terms relating to phase transitions of the solid state. Pure and Applied Chemistry, 66(3), 577-594. https://doi.org/10.1351/pac199466030577 DOI: 10.1351/pac199466030577 Document status and date: Published: 01/01/1994 Document Version: Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers) Please check the document version of this publication: • A submitted manuscript is the version of the article upon submission and before peer-review. There can be important differences between the submitted version and the official published version of record. People interested in the research are advised to contact the author for the final version of the publication, or visit the DOI to the publisher's website. • The final author version and the galley proof are versions of the publication after peer review. • The final published version features the final layout of the paper including the volume, issue and page numbers. Link to publication General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal. -

Notes on Phase Seperation Chem 130A Fall 2002 Prof
Notes on Phase Seperation Chem 130A Fall 2002 Prof. Groves Consider the process: Pure 1 + Pure 2 Æ Mixture of 1 and 2 We have previously derived the entropy of mixing for this process from statistical principles and found it to be: ∆ =− − Smix kN11ln X kN 2 ln X 2 where N1 and N2 are the number of molecules of type 1 and 2, respectively. We use k, the Boltzmann constant, since N1 and N2 are number of molecules. We could alternatively use R, the gas constant, if we chose to represent the number of molecules in terms of moles. X1 and X2 represent the mole fraction (e.g. X1 = N1 / (N1+N2)) of 1 and 2, respectively. If there are interactions between the two ∆ components, there will be a non-zero Hmix that we must consider. We can derive an expression ∆H = 2γ ∆ ∆ for Hmix using H of swapping one molecule of type 1, in pure type 1, with one molecule of type 2, in pure type 2 (see adjacent figure). We define γ as half of this ∆H of interchange. It is a differential interaction energy that tells us the energy difference between 1:1 and 2:2 interactions and 1:2 interactions. By defining 2γ = ∆H, γ represents the energy change associated with one molecule (note that two were involved in the swap). ∆ γ Now, to calculate Hmix from , we first consider 1 molecule of type 1 in the mixed system. For the moment we need not be concerned with whether or not they actually will mix, we just want ∆ to calculate Hmix as if they mix thoroughly. -

1000 J Mole Μa ( ) = 4,0001.09 TJ Mole Μb
3.012 PS 6 THERMODYANMICS SOLUTIONS 3.012 Issued: 11.28.05 Fall 2005 Due: THERMODYNAMICS 1. Building a binary phase diagram. Given below are data for a binary system of two materials A and B. The two components form three phases: an ideal liquid solution, and two solid phases α and β that exhibit regular solution behavior. Use the given thermodynamic data to answer the questions below. LIQUID PHASE: ALPHA PHASE: L J " J µo = "500 " 4.8T µo = #6000 ( A ) mole ( A ) mole L J " J µo = "1,000 "10T µo = #1,000 ( B ) mole ( B ) mole ! ! (T is temperature in K) Ω = 8,368 J/mole ! ! BETA PHASE: " J µo = #4,000 #1.09T ( A ) mole " J µo = #13,552 # 2.09T ( B ) mole ! (T is temperature in K) ! Ω = 7,000 J/mole a. Plot the free energy of L, alpha, and beta phases (overlaid on one plot) vs. XB at the following temperatures: 2000K, 1500K, 1000K, 925K, and 800K. For each plot, denote common tangents and drop vertical lines below the plot to a ‘composition bar’, as illustrated in the example plot below. For each graph, mark the stable phases as a function of XB in the composition bar below the graph, as illustrated in the example. 3.012 PS 6 1 of 18 12/11/05 EXAMPLE: 3.012 PS 6 2 of 18 12/11/05 3.012 PS 6 3 of 18 12/11/05 b. Using the free energy curves prepared in part (a), construct a qualitatively correct phase diagram for this system in the temperature range 800K – 2000K. -
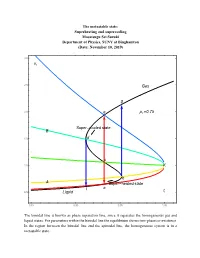
16.7 PT Superheating and Supercooling
The metastable state: Superheating and supercooling Masatsugu Sei Suzuki Department of Physics, SUNY at Binghamton (Date: November 10, 2019) 3.0 vr 2.5 Gas g 2.0 e pr 0.75 Super cooled state B 1.5 d c 1.0 K b A Super heated state l a 0.5 Liquid tr 0.85 0.90 0.95 1.00 The binodal line is known as phase separation line, since it separates the homogeneous gas and liquid states. For parameters within the binodal line the equilibrium shows two-phase co-existence. In the region between the binodal line and the spinodal line, the homogeneous system is in a metastable state. 3.0 vr 2.5 Gas p 0.70 r g pr 0.75 2.0 e pr 0.80 pr 0.85 B pr 0.90 1.5 d pr 0.95 c 1.0 K b A a l t 0.5 Liquid r 0.85 0.90 0.95 1.00 3.0 vr 2.5 Gas 2.0 g pr 0.80 e Super cooled state B 1.5 d c 1.0 K b A a l Super heated state 0.5 Liquid tr 0.85 0.90 0.95 1.00 e: the intersection of the ParametricPlot (denoted by black line) and the K-e line (binodal line) d: the intersection of the ParametricPlot (denoted by black line) and the K-d line (spinodal line) c: the intersection of the ParametricPlot (denoted by black line) and the K-c line b: the intersection of the ParametricPlot (denoted by black line) and the K-b line (spinodal line) a: the intersection of the ParametricPlot (denoted by black line) and the K-a line (binodal line) Point a: thermal equilibrium Point e: thermal equilibrium Point c: intermediate between the points a and b (temperature of point c is the same as that of points and e). -
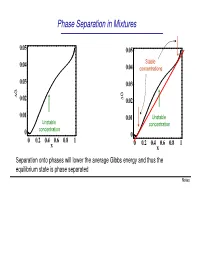
Phase Separation in Mixtures
Phase Separation in Mixtures 0.05 0.05 0.04 Stable 0.04 concentrations 0.03 0.03 G G ∆ 0.02 ∆ 0.02 0.01 0.01 Unstable Unstable concentration concentration 0 0 0 0.2 0.4 0.6 0.8 1 0 0.2 0.4 0.6 0.8 1 x x Separation onto phases will lower the average Gibbs energy and thus the equilibrium state is phase separated Notes Phase Separation versus Temperature Note that at higher temperatures the region of concentrations where phase separation takes place shrinks and G eventually disappears T increases ∆G = ∆U −T∆S This is because the term -T∆S becomes large at high temperatures. You can say that entropy “wins” over the potential energy cost at high temperatures Microscopically, the kinetic energy becomes x much larger than potential at high T, and the 1 x2 molecules randomly “run around” without noticing potential energy and thus intermix Oil and water mix at high temperature Notes Liquid-Gas Phase Separation in a Mixture A binary mixture can exist in liquid or gas phases. The liquid and gas phases have different Gibbs potentials as a function of mole fraction of one of the components of the mixture At high temperatures the Ggas <Gliquid because the entropy of gas is greater than that of liquid Tb2 T At lower temperatures, the two Gibbs b1 potentials intersect and separation onto gas and liquid takes place. Notes Boiling of a binary mixture Bubble point curve Dew point If we have a liquid at pint A that contains F A gas curve a molar fraction x1 of component 1 and T start heating it: E D - First the liquid heats to a temperature T’ C at constant x A and arrives to point B. -

Phase Equilibria of Quasi-Ternary Systems Consisting of Multicomponent Polymers in a Binary Solvent Mixture IV
Polymer Journal, Vol. 18, No.4, pp 347-360 (1986) Phase Equilibria of Quasi-Ternary Systems Consisting of Multicomponent Polymers in a Binary Solvent Mixture IV. Spinodal Curve and Critical Solution Point Kenji KAMIDE and Shigenobu MATSUDA Fundamental Research Laboratory of Fiber and Fiber-Forming Polymers, Asahi Chemical Industry Company, Ltd., 11-7, Hacchonawate, Takatsuki, Osaka 569, Japan (Received October 28, 1985) ABSTRACT: An attempt was made to construct a quasi-ternary system cons1stmg of multicomponent polymers dissolved in binary solvent mixture (solvents I and 2) to derive the equations of spinodal and neutral equilibrium conditions and to clarify the effects of three thermodynamic interaction x-parameters between solvent I and solvent 2, solvent I and polymer, and solvent 2 and polymer(x 12, x13, and x23) and the weight-average degree of polymerization and the ratio of to the number-average degree of polymerization of the original polymer on the spinodal curve and the critical points (the critical concentration calculated from the above mentioned equations. The cloud point curve was also calculated indirectly from coexisting curve evaluated according to a method in our previous papers and constant (starting polymer volume fraction) line. The cross point of a constant line and a coexisting curve is a cloud point. The cloud point curve thus calculated was confirmed to coincide, as theory predicted with the spinodal curve, at the critical point. decreases with an increase in X12, x23, and and increases with an increase in x13