MSE Problems
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Recent Progress on Deep Eutectic Solvents in Biocatalysis Pei Xu1,2, Gao‑Wei Zheng3, Min‑Hua Zong1,2, Ning Li1 and Wen‑Yong Lou1,2*
Xu et al. Bioresour. Bioprocess. (2017) 4:34 DOI 10.1186/s40643-017-0165-5 REVIEW Open Access Recent progress on deep eutectic solvents in biocatalysis Pei Xu1,2, Gao‑Wei Zheng3, Min‑Hua Zong1,2, Ning Li1 and Wen‑Yong Lou1,2* Abstract Deep eutectic solvents (DESs) are eutectic mixtures of salts and hydrogen bond donors with melting points low enough to be used as solvents. DESs have proved to be a good alternative to traditional organic solvents and ionic liq‑ uids (ILs) in many biocatalytic processes. Apart from the benign characteristics similar to those of ILs (e.g., low volatil‑ ity, low infammability and low melting point), DESs have their unique merits of easy preparation and low cost owing to their renewable and available raw materials. To better apply such solvents in green and sustainable chemistry, this review frstly describes some basic properties, mainly the toxicity and biodegradability of DESs. Secondly, it presents several valuable applications of DES as solvent/co-solvent in biocatalytic reactions, such as lipase-catalyzed transester‑ ifcation and ester hydrolysis reactions. The roles, serving as extractive reagent for an enzymatic product and pretreat‑ ment solvent of enzymatic biomass hydrolysis, are also discussed. Further understanding how DESs afect biocatalytic reaction will facilitate the design of novel solvents and contribute to the discovery of new reactions in these solvents. Keywords: Deep eutectic solvents, Biocatalysis, Catalysts, Biodegradability, Infuence Introduction (1998), one concept is “Safer Solvents and Auxiliaries”. Biocatalysis, defned as reactions catalyzed by bio- Solvents represent a permanent challenge for green and catalysts such as isolated enzymes and whole cells, has sustainable chemistry due to their vast majority of mass experienced signifcant progress in the felds of either used in catalytic processes (Anastas and Eghbali 2010). -

Phase Diagrams and Phase Separation
Phase Diagrams and Phase Separation Books MF Ashby and DA Jones, Engineering Materials Vol 2, Pergamon P Haasen, Physical Metallurgy, G Strobl, The Physics of Polymers, Springer Introduction Mixing two (or more) components together can lead to new properties: Metal alloys e.g. steel, bronze, brass…. Polymers e.g. rubber toughened systems. Can either get complete mixing on the atomic/molecular level, or phase separation. Phase Diagrams allow us to map out what happens under different conditions (specifically of concentration and temperature). Free Energy of Mixing Entropy of Mixing nA atoms of A nB atoms of B AM Donald 1 Phase Diagrams Total atoms N = nA + nB Then Smix = k ln W N! = k ln nA!nb! This can be rewritten in terms of concentrations of the two types of atoms: nA/N = cA nB/N = cB and using Stirling's approximation Smix = -Nk (cAln cA + cBln cB) / kN mix S AB0.5 This is a parabolic curve. There is always a positive entropy gain on mixing (note the logarithms are negative) – so that entropic considerations alone will lead to a homogeneous mixture. The infinite slope at cA=0 and 1 means that it is very hard to remove final few impurities from a mixture. AM Donald 2 Phase Diagrams This is the situation if no molecular interactions to lead to enthalpic contribution to the free energy (this corresponds to the athermal or ideal mixing case). Enthalpic Contribution Assume a coordination number Z. Within a mean field approximation there are 2 nAA bonds of A-A type = 1/2 NcAZcA = 1/2 NZcA nBB bonds of B-B type = 1/2 NcBZcB = 1/2 NZ(1- 2 cA) and nAB bonds of A-B type = NZcA(1-cA) where the factor 1/2 comes in to avoid double counting and cB = (1-cA). -

Thermodynamics and Kinetics of Nucleation in Binary Solutions
1 Thermodynamics and kinetics of nucleation in binary solutions Nikolay V. Alekseechkin Akhiezer Institute for Theoretical Physics, National Science Centre “Kharkov Institute of Physics and Technology”, Akademicheskaya Street 1, Kharkov 61108, Ukraine Email: [email protected] Keywords: Binary solution; phase equilibrium; binary nucleation; diffusion growth; surface layer. A new approach that is a combination of classical thermodynamics and macroscopic kinetics is offered for studying the nucleation kinetics in condensed binary solutions. The theory covers the separation of liquid and solid solutions proceeding along the nucleation mechanism, as well as liquid-solid transformations, e.g., the crystallization of molten alloys. The cases of nucleation of both unary and binary precipitates are considered. Equations of equilibrium for a critical nucleus are derived and then employed in the macroscopic equations of nucleus growth; the steady state nucleation rate is calculated with the use of these equations. The present approach can be applied to the general case of non-ideal solution; the calculations are performed on the model of regular solution within the classical nucleation theory (CNT) approximation implying the bulk properties of a nucleus and constant surface tension. The way of extending the theory beyond the CNT approximation is shown in the framework of the finite- thickness layer method. From equations of equilibrium of a surface layer with coexisting bulk phases, equations for adsorption and the dependences of surface tension on temperature, radius, and composition are derived. Surface effects on the thermodynamics and kinetics of nucleation are discussed. 1. Introduction Binary nucleation covers a wide class of processes of phase transformations which can be divided into three groups: (i) gas-liquid (or solid) transformations, (ii) liquid-gas transformations, and (iii) transformations within a condensed state. -

Phase Diagrams
Module-07 Phase Diagrams Contents 1) Equilibrium phase diagrams, Particle strengthening by precipitation and precipitation reactions 2) Kinetics of nucleation and growth 3) The iron-carbon system, phase transformations 4) Transformation rate effects and TTT diagrams, Microstructure and property changes in iron- carbon system Mixtures – Solutions – Phases Almost all materials have more than one phase in them. Thus engineering materials attain their special properties. Macroscopic basic unit of a material is called component. It refers to a independent chemical species. The components of a system may be elements, ions or compounds. A phase can be defined as a homogeneous portion of a system that has uniform physical and chemical characteristics i.e. it is a physically distinct from other phases, chemically homogeneous and mechanically separable portion of a system. A component can exist in many phases. E.g.: Water exists as ice, liquid water, and water vapor. Carbon exists as graphite and diamond. Mixtures – Solutions – Phases (contd…) When two phases are present in a system, it is not necessary that there be a difference in both physical and chemical properties; a disparity in one or the other set of properties is sufficient. A solution (liquid or solid) is phase with more than one component; a mixture is a material with more than one phase. Solute (minor component of two in a solution) does not change the structural pattern of the solvent, and the composition of any solution can be varied. In mixtures, there are different phases, each with its own atomic arrangement. It is possible to have a mixture of two different solutions! Gibbs phase rule In a system under a set of conditions, number of phases (P) exist can be related to the number of components (C) and degrees of freedom (F) by Gibbs phase rule. -

Interval Mathematics Applied to Critical Point Transitions
Revista de Matematica:´ Teor´ıa y Aplicaciones 2005 12(1 & 2) : 29–44 cimpa – ucr – ccss issn: 1409-2433 interval mathematics applied to critical point transitions Benito A. Stradi∗ Received/Recibido: 16 Feb 2004 Abstract The determination of critical points of mixtures is important for both practical and theoretical reasons in the modeling of phase behavior, especially at high pressure. The equations that describe the behavior of complex mixtures near critical points are highly nonlinear and with multiplicity of solutions to the critical point equations. Interval arithmetic can be used to reliably locate all the critical points of a given mixture. The method also verifies the nonexistence of a critical point if a mixture of a given composition does not have one. This study uses an interval Newton/Generalized Bisection algorithm that provides a mathematical and computational guarantee that all mixture critical points are located. The technique is illustrated using several ex- ample problems. These problems involve cubic equation of state models; however, the technique is general purpose and can be applied in connection with other nonlinear problems. Keywords: Critical Points, Interval Analysis, Computational Methods. Resumen La determinaci´onde puntos cr´ıticosde mezclas es importante tanto por razones pr´acticascomo te´oricasen el modelamiento del comportamiento de fases, especial- mente a presiones altas. Las ecuaciones que describen el comportamiento de mezclas complejas cerca del punto cr´ıticoson significativamente no lineales y con multipli- cidad de soluciones para las ecuaciones del punto cr´ıtico. Aritm´eticade intervalos puede ser usada para localizar con confianza todos los puntos cr´ıticosde una mezcla dada. -

Determination of Supercooling Degree, Nucleation and Growth Rates, and Particle Size for Ice Slurry Crystallization in Vacuum
crystals Article Determination of Supercooling Degree, Nucleation and Growth Rates, and Particle Size for Ice Slurry Crystallization in Vacuum Xi Liu, Kunyu Zhuang, Shi Lin, Zheng Zhang and Xuelai Li * School of Chemical Engineering, Fuzhou University, Fuzhou 350116, China; [email protected] (X.L.); [email protected] (K.Z.); [email protected] (S.L.); [email protected] (Z.Z.) * Correspondence: [email protected] Academic Editors: Helmut Cölfen and Mei Pan Received: 7 April 2017; Accepted: 29 April 2017; Published: 5 May 2017 Abstract: Understanding the crystallization behavior of ice slurry under vacuum condition is important to the wide application of the vacuum method. In this study, we first measured the supercooling degree of the initiation of ice slurry formation under different stirring rates, cooling rates and ethylene glycol concentrations. Results indicate that the supercooling crystallization pressure difference increases with increasing cooling rate, while it decreases with increasing ethylene glycol concentration. The stirring rate has little influence on supercooling crystallization pressure difference. Second, the crystallization kinetics of ice crystals was conducted through batch cooling crystallization experiments based on the population balance equation. The equations of nucleation rate and growth rate were established in terms of power law kinetic expressions. Meanwhile, the influences of suspension density, stirring rate and supercooling degree on the process of nucleation and growth were studied. Third, the morphology of ice crystals in ice slurry was obtained using a microscopic observation system. It is found that the effect of stirring rate on ice crystal size is very small and the addition of ethylene glycoleffectively inhibits the growth of ice crystals. -
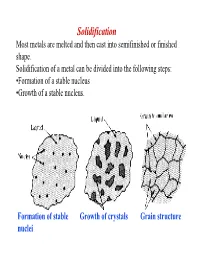
Solidification Most Metals Are Melted and Then Cast Into Semifinished Or Finished Shape
Solidification Most metals are melted and then cast into semifinished or finished shape. Solidification of a metal can be divided into the following steps: •Formation of a stable nucleus •Growth of a stable nucleus. Formation of stable Growth of crystals Grain structure nuclei Polycrystalline Metals In most cases, solidification begins from multiple sites, each of which can produce a different orientation The result is a “polycrystalline” material consisting of many small crystals of “grains” Each grain has the same crystal lattice, but the lattices are misoriented from grain to grain Driving force: solidification ⇒ For the reaction to proceed to the right ∆G AL AS V must be negative. • Writing the free energies of the solid and liquid as: S S S GV = H - TS L L L GV = H - TS ∴∴∴ ∆GV = ∆H - T∆S • At equilibrium, i.e. Tmelt , then the ∆GV = 0 , so we can estimate the melting entropy as: ∆S = ∆H/Tmelt where -∆H is the latent heat (enthalpy) of melting. • Ignore the difference in specific heat between solid and liquid, and we estimate the free energy difference as: T ∆H × ∆T ∆GV ≅ ∆H − ∆H = TMelt TMelt NUCLEATION The two main mechanisms by which nucleation of a solid particles in liquid metal occurs are homogeneous and heterogeneous nucleation. Homogeneous Nucleation Homogeneous nucleation occurs when there are no special objects inside a phase which can cause nucleation. For instance when a pure liquid metal is slowly cooled below its equilibrium freezing temperature to a sufficient degree numerous homogeneous nuclei are created by slow-moving atoms bonding together in a crystalline form. -

Study of the LLE, VLE and VLLE of the Ternary System Water + 1-Butanol + Isoamyl Alcohol at 101.3 Kpa
View metadata, citation and similar papers at core.ac.uk brought to you by CORE Submitted to Journal of Chemical & Engineering Data provided by Repositorio Institucional de la Universidad de Alicante This document is confidential and is proprietary to the American Chemical Society and its authors. Do not copy or disclose without written permission. If you have received this item in error, notify the sender and delete all copies. Study of the LLE, VLE and VLLE of the ternary system water + 1-butanol + isoamyl alcohol at 101.3 kPa Journal: Journal of Chemical & Engineering Data Manuscript ID je-2018-00308r.R3 Manuscript Type: Article Date Submitted by the Author: 27-Aug-2018 Complete List of Authors: Saquete, María Dolores; Universitat d'Alacant, Chemical Engineering Font, Alicia; Universitat d'Alacant, Chemical Engineering Garcia-Cano, Jorge; Universitat d'Alacant, Chemical Engineering Blasco, Inmaculada; Universitat d'Alacant, Chemical Engineering ACS Paragon Plus Environment Page 1 of 21 Submitted to Journal of Chemical & Engineering Data 1 2 3 Study of the LLE, VLE and VLLE of the ternary system water + 4 1-butanol + isoamyl alcohol at 101.3 kPa 5 6 María Dolores Saquete, Alicia Font, Jorge García-Cano* and Inmaculada Blasco. 7 8 University of Alicante, P.O. Box 99, E-03080 Alicante, Spain 9 10 Abstract 11 12 In this work it has been determined experimentally the liquidliquid equilibrium of the 13 water + 1butanol + isoamyl alcohol system at 303.15K and 313.15K. The UNIQUAC, 14 NRTL and UNIFAC models have been employed to correlate and predict LLE and 15 16 compare them with the experimental data. -

Chapter 9: Other Topics in Phase Equilibria
Chapter 9: Other Topics in Phase Equilibria This chapter deals with relations that derive in cases of equilibrium between combinations of two co-existing phases other than vapour and liquid, i.e., liquid-liquid, solid-liquid, and solid- vapour. Each of these phase equilibria may be employed to overcome difficulties encountered in purification processes that exploit the difference in the volatilities of the components of a mixture, i.e., by vapour-liquid equilibria. As with the case of vapour-liquid equilibria, the objective is to derive relations that connect the compositions of the two co-existing phases as functions of temperature and pressure. 9.1 Liquid-liquid Equilibria (LLE) 9.1.1 LLE Phase Diagrams Unlike gases which are miscible in all proportions at low pressures, liquid solutions (binary or higher order) often display partial immiscibility at least over certain range of temperature, and composition. If one attempts to form a solution within that certain composition range the system splits spontaneously into two liquid phases each comprising a solution of different composition. Thus, in such situations the equilibrium state of the system is two phases of a fixed composition corresponding to a temperature. The compositions of two such phases, however, change with temperature. This typical phase behavior of such binary liquid-liquid systems is depicted in fig. 9.1a. The closed curve represents the region where the system exists Fig. 9.1 Phase diagrams for a binary liquid system showing partial immiscibility in two phases, while outside it the state is a homogenous single liquid phase. Take for example, the point P (or Q). -

Section 1 Introduction to Alloy Phase Diagrams
Copyright © 1992 ASM International® ASM Handbook, Volume 3: Alloy Phase Diagrams All rights reserved. Hugh Baker, editor, p 1.1-1.29 www.asminternational.org Section 1 Introduction to Alloy Phase Diagrams Hugh Baker, Editor ALLOY PHASE DIAGRAMS are useful to exhaust system). Phase diagrams also are con- terms "phase" and "phase field" is seldom made, metallurgists, materials engineers, and materials sulted when attacking service problems such as and all materials having the same phase name are scientists in four major areas: (1) development of pitting and intergranular corrosion, hydrogen referred to as the same phase. new alloys for specific applications, (2) fabrica- damage, and hot corrosion. Equilibrium. There are three types of equili- tion of these alloys into useful configurations, (3) In a majority of the more widely used commer- bria: stable, metastable, and unstable. These three design and control of heat treatment procedures cial alloys, the allowable composition range en- conditions are illustrated in a mechanical sense in for specific alloys that will produce the required compasses only a small portion of the relevant Fig. l. Stable equilibrium exists when the object mechanical, physical, and chemical properties, phase diagram. The nonequilibrium conditions is in its lowest energy condition; metastable equi- and (4) solving problems that arise with specific that are usually encountered inpractice, however, librium exists when additional energy must be alloys in their performance in commercial appli- necessitate the knowledge of a much greater por- introduced before the object can reach true stabil- cations, thus improving product predictability. In tion of the diagram. Therefore, a thorough under- ity; unstable equilibrium exists when no addi- all these areas, the use of phase diagrams allows standing of alloy phase diagrams in general and tional energy is needed before reaching meta- research, development, and production to be done their practical use will prove to be of great help stability or stability. -

Definitions of Terms Relating to Phase Transitions of the Solid State
Definitions of terms relating to phase transitions of the solid state Citation for published version (APA): Clark, J. B., Hastie, J. W., Kihlborg, L. H. E., Metselaar, R., & Thackeray, M. M. (1994). Definitions of terms relating to phase transitions of the solid state. Pure and Applied Chemistry, 66(3), 577-594. https://doi.org/10.1351/pac199466030577 DOI: 10.1351/pac199466030577 Document status and date: Published: 01/01/1994 Document Version: Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers) Please check the document version of this publication: • A submitted manuscript is the version of the article upon submission and before peer-review. There can be important differences between the submitted version and the official published version of record. People interested in the research are advised to contact the author for the final version of the publication, or visit the DOI to the publisher's website. • The final author version and the galley proof are versions of the publication after peer review. • The final published version features the final layout of the paper including the volume, issue and page numbers. Link to publication General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal. -

Notes on Phase Seperation Chem 130A Fall 2002 Prof
Notes on Phase Seperation Chem 130A Fall 2002 Prof. Groves Consider the process: Pure 1 + Pure 2 Æ Mixture of 1 and 2 We have previously derived the entropy of mixing for this process from statistical principles and found it to be: ∆ =− − Smix kN11ln X kN 2 ln X 2 where N1 and N2 are the number of molecules of type 1 and 2, respectively. We use k, the Boltzmann constant, since N1 and N2 are number of molecules. We could alternatively use R, the gas constant, if we chose to represent the number of molecules in terms of moles. X1 and X2 represent the mole fraction (e.g. X1 = N1 / (N1+N2)) of 1 and 2, respectively. If there are interactions between the two ∆ components, there will be a non-zero Hmix that we must consider. We can derive an expression ∆H = 2γ ∆ ∆ for Hmix using H of swapping one molecule of type 1, in pure type 1, with one molecule of type 2, in pure type 2 (see adjacent figure). We define γ as half of this ∆H of interchange. It is a differential interaction energy that tells us the energy difference between 1:1 and 2:2 interactions and 1:2 interactions. By defining 2γ = ∆H, γ represents the energy change associated with one molecule (note that two were involved in the swap). ∆ γ Now, to calculate Hmix from , we first consider 1 molecule of type 1 in the mixed system. For the moment we need not be concerned with whether or not they actually will mix, we just want ∆ to calculate Hmix as if they mix thoroughly.