Nucleation Theory: a Literature Review and Applications to Nucleation Rates of Natural Gas Hydrates
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Thermodynamics and Kinetics of Nucleation in Binary Solutions
1 Thermodynamics and kinetics of nucleation in binary solutions Nikolay V. Alekseechkin Akhiezer Institute for Theoretical Physics, National Science Centre “Kharkov Institute of Physics and Technology”, Akademicheskaya Street 1, Kharkov 61108, Ukraine Email: [email protected] Keywords: Binary solution; phase equilibrium; binary nucleation; diffusion growth; surface layer. A new approach that is a combination of classical thermodynamics and macroscopic kinetics is offered for studying the nucleation kinetics in condensed binary solutions. The theory covers the separation of liquid and solid solutions proceeding along the nucleation mechanism, as well as liquid-solid transformations, e.g., the crystallization of molten alloys. The cases of nucleation of both unary and binary precipitates are considered. Equations of equilibrium for a critical nucleus are derived and then employed in the macroscopic equations of nucleus growth; the steady state nucleation rate is calculated with the use of these equations. The present approach can be applied to the general case of non-ideal solution; the calculations are performed on the model of regular solution within the classical nucleation theory (CNT) approximation implying the bulk properties of a nucleus and constant surface tension. The way of extending the theory beyond the CNT approximation is shown in the framework of the finite- thickness layer method. From equations of equilibrium of a surface layer with coexisting bulk phases, equations for adsorption and the dependences of surface tension on temperature, radius, and composition are derived. Surface effects on the thermodynamics and kinetics of nucleation are discussed. 1. Introduction Binary nucleation covers a wide class of processes of phase transformations which can be divided into three groups: (i) gas-liquid (or solid) transformations, (ii) liquid-gas transformations, and (iii) transformations within a condensed state. -

Determination of Supercooling Degree, Nucleation and Growth Rates, and Particle Size for Ice Slurry Crystallization in Vacuum
crystals Article Determination of Supercooling Degree, Nucleation and Growth Rates, and Particle Size for Ice Slurry Crystallization in Vacuum Xi Liu, Kunyu Zhuang, Shi Lin, Zheng Zhang and Xuelai Li * School of Chemical Engineering, Fuzhou University, Fuzhou 350116, China; [email protected] (X.L.); [email protected] (K.Z.); [email protected] (S.L.); [email protected] (Z.Z.) * Correspondence: [email protected] Academic Editors: Helmut Cölfen and Mei Pan Received: 7 April 2017; Accepted: 29 April 2017; Published: 5 May 2017 Abstract: Understanding the crystallization behavior of ice slurry under vacuum condition is important to the wide application of the vacuum method. In this study, we first measured the supercooling degree of the initiation of ice slurry formation under different stirring rates, cooling rates and ethylene glycol concentrations. Results indicate that the supercooling crystallization pressure difference increases with increasing cooling rate, while it decreases with increasing ethylene glycol concentration. The stirring rate has little influence on supercooling crystallization pressure difference. Second, the crystallization kinetics of ice crystals was conducted through batch cooling crystallization experiments based on the population balance equation. The equations of nucleation rate and growth rate were established in terms of power law kinetic expressions. Meanwhile, the influences of suspension density, stirring rate and supercooling degree on the process of nucleation and growth were studied. Third, the morphology of ice crystals in ice slurry was obtained using a microscopic observation system. It is found that the effect of stirring rate on ice crystal size is very small and the addition of ethylene glycoleffectively inhibits the growth of ice crystals. -
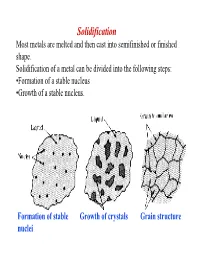
Solidification Most Metals Are Melted and Then Cast Into Semifinished Or Finished Shape
Solidification Most metals are melted and then cast into semifinished or finished shape. Solidification of a metal can be divided into the following steps: •Formation of a stable nucleus •Growth of a stable nucleus. Formation of stable Growth of crystals Grain structure nuclei Polycrystalline Metals In most cases, solidification begins from multiple sites, each of which can produce a different orientation The result is a “polycrystalline” material consisting of many small crystals of “grains” Each grain has the same crystal lattice, but the lattices are misoriented from grain to grain Driving force: solidification ⇒ For the reaction to proceed to the right ∆G AL AS V must be negative. • Writing the free energies of the solid and liquid as: S S S GV = H - TS L L L GV = H - TS ∴∴∴ ∆GV = ∆H - T∆S • At equilibrium, i.e. Tmelt , then the ∆GV = 0 , so we can estimate the melting entropy as: ∆S = ∆H/Tmelt where -∆H is the latent heat (enthalpy) of melting. • Ignore the difference in specific heat between solid and liquid, and we estimate the free energy difference as: T ∆H × ∆T ∆GV ≅ ∆H − ∆H = TMelt TMelt NUCLEATION The two main mechanisms by which nucleation of a solid particles in liquid metal occurs are homogeneous and heterogeneous nucleation. Homogeneous Nucleation Homogeneous nucleation occurs when there are no special objects inside a phase which can cause nucleation. For instance when a pure liquid metal is slowly cooled below its equilibrium freezing temperature to a sufficient degree numerous homogeneous nuclei are created by slow-moving atoms bonding together in a crystalline form. -

Crystallization of Supercooled Liquid Elements Induced by Superclusters Containing Magic Atom Numbers Robert F
Crystallization of Supercooled Liquid Elements Induced by Superclusters Containing Magic Atom Numbers Robert F. Tournier, CRETA /CNRS, Université Joseph Fourier, B.P. 166, 38042 Grenoble cedex 09, France. E-mail: [email protected]; Tel.: +33-608-716-878; Fax: +33-956-705-473. Abstract: A few experiments have detected icosahedral superclusters in undercooled liquids. These superclusters survive above the crystal melting temperature Tm because all their surface atoms have the same fusion heat as their core atoms and are melted by liquid homogeneous and heterogeneous nucleation in their core, depending on superheating time and temperature. They act as heterogeneous growth nuclei of crystallized phase at a temperature Tc of the undercooled melt. They contribute to the critical barrier reduction, which becomes smaller than that of crystals containing the same atom number n. After strong superheating, the undercooling rate is still limited because the nucleation of 13-atom superclusters always reduces this barrier, and increases Tc above a homogeneous nucleation temperature equal to Tm/3 in liquid elements. After weak superheating, the most stable superclusters containing n = 13, 55, 147, 309 and 561 atoms survive or melt and determine Tc during undercooling, depending on n and sample volume. The experimental nucleation temperatures Tc of 32 liquid elements and the supercluster melting temperatures are predicted with sample volumes varying by 18 orders of magnitude. The classical Gibbs free energy change is used, adding an enthalpy saving related to the Laplace pressure change associated with supercluster formation, which is quantified for n=13 and 55. Keywords: thermal properties, solid-liquid interface energy, crystal nucleation, undercooling, superclusters, liquid-solid transition, overheating, non-metal to metal transition in cluster, Laplace pressure 1. -

Comparison of Classical Nucleation Theory and Modern Theory of Phase
ed Chem nc i a ca Baranov, J Adv Chem Eng 2017, 7:1 v l d E A n f g DOI: 10.4172/2090-4568.1000177 o i n l e Journal of Advanced a e n r r i n u g o J ISSN: 2090-4568 Chemical Engineering Research Article Open Access Comparison of Classical Nucleation Theory and Modern Theory of Phase Transition Baranov SA1,2,3* 1Institute of Applied Physics, Academy of Sciences of Moldova, Republic of Moldova 2Department de Genie Physique, Ecole Polytechnique de Montreal, Montreal H3C 3A7, Quebec, Canada 3Shevchenko Transnistria State University, Tiraspol, Republic of Moldova Abstract The formation of electrochemical phase is a typical example of a first-order phase transition. An overview on old and new thermodynamic and kinetic aspects of the theoretical description of first-order phase transitions, and, in particular, of the theory of nucleation is given. Electrochemical nucleation of nanostructures has been considered in terms of the classical Gibbs and the Cahn-Hilliard-Hillert theories. We obtained agreement between the theories Kahn-Hilliard-Hillert and Gibbs. Keywords: Electrochemistry; Nucleation; Nanoparticles; Phase rc=2γ/μ transitions; Gibbs theory; Cahn-Hilliard theory Critical nucleus radius corresponding to maximum G3 is as follows Introduction (Figure 1) [1-9]: 32 (5) Electrochemical nucleation is usually described in the framework ∆G3 =(16π / 3)( γµ33 / ) of the classical Gibbs theory of nucleation, which provides a simple For a two-dimensional case, we obtain [1-9] expression for the critical radius of a growing nucleus (nanoparticles) 2 [1-11]: ∆=G2 =G 2P / 2πγ (2 / µ2 ) where γ2 is the specific surface energy (for the two-dimensional rc=К(γ/μ) 1 case), μ3 is the change in volume energy during a phase transition (for where γ is the specific surface energy, μ is the change in volume the two-dimensional case). -

Origins of the Failure of Classical Nucleation Theory for Nanocellular Polymer Foams
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by University of Waterloo's Institutional Repository Origins of the Failure of Classical Nucleation Theory for Nanocellular Polymer Foams Yeongyoon Kim,1 Chul B. Park,2 P. Chen,3, 4 and Russell B. Thompson1,4, ∗ 1Department of Physics and Astronomy, University of Waterloo, 200 University Avenue West, Waterloo, Ontario, Canada N2L 3G1 2Microcellular Plastics Manufacturing Laboratory, Department of Mechanical and Industrial Engineering, University of Toronto, 5 King’s College Road, Toronto, Ontario, Canada M5S 3G8 3Department of Chemical Engineering, University of Waterloo, 200 University Avenue West, Waterloo, Ontario, Canada N2L 3G1 4Waterloo Institute for Nanotechnology, University of Waterloo, 200 University Avenue West, Waterloo, Ontario, Canada N2L 3G1 This document is the accepted manuscript version of a published article. Published by The Royal Society of Chemistry in the journal "Soft Matter" issue 16, DOI: 10.1039/C1SM05575E 1 Abstract Relative nucleation rates for fluid bubbles of nanometre dimensions in polymer matrices are calculated using both classical nucleation theory and self-consistent field theory. An identical model is used for both calculations showing that classical nucleation theory predictions are off by many orders of magnitude. The main cause of the failure of classical nucleation theory can be traced to its representation of a bubble surface as a flat interface. For nanoscopic bubbles, the curvature of the bubble surface is comparable to the size of the polymer molecules. Polymers on the outside of a curved bubble surface can explore more conformations than can polymers next to a flat interface. -

Applications
PHY474 Applications Ulrich Z¨urcher∗ Physics Department, Cleveland State University, Cleveland, OH 44115 (Dated: November 14, 2008) PACS numbers: 05.70 I. PHASE TRANSFORMATION OF PURE not coexist (in equilibrium): ice directy sublimates into SUBSTANCES vapor. Sublimation of dry ice [carbondioxide]. the triple point lies above atmospheric pressure. Most substances: The curve of pressure vs volume for a quantity of applying more pressure raises the melting temperature. matter at constant temperature is determined by the Ice is different: applying more pressure lowers its melting Gibbs free energy of the substance. The curve is called temperature: this is related to the fact that density of ice isotherm. We consider the isotherms of a real gas in is lower than density of liquid. which atoms associate together in a liquid or solid phase. Liquid-gas phase boundary always has a poisitive A phase is a prortion of the system that is uniform in slope: if you increase temperature you must increase composition. pressure to prevent liquid from vaporizing. As pressure Two phases may coexist with a definite boundary be- increases, density of gas is increasing so gas and liquid tween them. An isotherm may show a region in the PV become indistinguishable. Eventually there is a point when there is no longer a discontinuous transition: crit- diagram in which liquid and gas coexist in equilibrium ◦ with each other. There are isotherms at low tempera- ical point. For water it is 374 C and 221 bars. Close to tures for which solids and liquids coexist and isotherms the critical point we call the system “fluid." for which solids and gases coexist. -

Introduction to the Physics of Nucleation
C. R. Physique 7 (2006) 946–958 http://france.elsevier.com/direct/COMREN/ Nucleation/Nucléation Introduction to the physics of nucleation Humphrey J. Maris Department of Physics, Brown University, Providence, RI 02912, USA Available online 28 November 2006 Abstract In this introductory article, I review the theory of nucleation by thermal activation and by quantum tunneling. The effect of heterogeneous nucleation at surfaces is discussed and a brief survey of experimental techniques is given. To cite this article: H.J. Maris, C. R. Physique 7 (2006). © 2006 Académie des sciences. Published by Elsevier Masson SAS. All rights reserved. Résumé Introduction à la physique de la nucléation. Dans cet article introductif, je passe en revue la théorie de la nucléation par activation thermique et par effet tunnel quantique. Je discute les effets de la nucléation hétérogène aux surfaces et je propose un bref survol des techniques expérimentales utilisées. Pour citer cet article : H.J. Maris, C. R. Physique 7 (2006). © 2006 Académie des sciences. Published by Elsevier Masson SAS. All rights reserved. Keywords: Nucleation; Phase transitions Mots-clés : Nucleation ; Transitions de phase This issue contains a number of reviews all concerning different aspects of nucleation. In this introduction, I briefly summarize the basic theory of nucleation processes and then mention some selected topics of interest. Nucleation means a change in a physical or chemical system that begins within a small region, that is to say, that begins at a nucleus. The very long history of this subject has been reviewed by G.S. Kell [1] who credits Huyghens with having in 1662 performed the first experiments with metastable water under tension. -

MSE Problems
Materials Science and Engineering Problems MSE Faculty April 19, 2021 This document includes the assigned problems that have been included so far in the digitized portion of our course curriculum. Contents 1 301 Problems 3 1.1 Course Organization.................................................. 3 1.2 Atomic Structure and Bonding ............................................ 3 1.3 Crystal Structure .................................................... 5 1.4 Imperfections ...................................................... 8 1.5 Electrical Properties................................................... 10 1.6 Diffusion......................................................... 12 1.7 Phase Diagrams..................................................... 14 1.8 Phase Transformations................................................. 18 1.9 Mechanical Properties ................................................. 18 1.10 Fracture Mechanics................................................... 22 1.11 Corrosion......................................................... 23 1.12 Ceramics......................................................... 24 1.13 Polymers......................................................... 25 2 314 Problems 27 2.1 Phases and Components................................................ 27 2.2 Intensive and Extensive Properties.......................................... 27 2.3 Differential Quantities and State Functions ..................................... 27 2.4 Entropy.......................................................... 27 2.5 -

Chapter 10: Phase Transformations
Chapter 10: Phase Transformations ISSUES TO ADDRESS... • Transforming one phase into another takes time. Fe Fe C Eutectoid 3 g transformation (cementite) (Austenite) + a C FCC (ferrite) (BCC) • How does the rate of transformation depend on time and T? • How can we slow down the transformation so that we can engineering non-equilibrium structures? • Are the mechanical properties of non-equilibrium structures better? Chapter 10 - 1 Phase transformation • Takes time (transformation rates: kinetics). • Involves movement/rearrangement of atoms. • Usually involves changes in microstructure 1. “Simple” diffusion-dependent transformation: no change in number of compositions of phases present (e.g. solidification of pure elemental metals, allotropic transformation, recrystallization, grain growth). 2. Diffusion-dependent transformation: transformation with alteration in phase composition and, often, with changes in number of phases present (e.g. eutectoid reaction). 3. Diffusionless transformation: e.g. rapid T quenching to “trap” metastable phases. Chapter 10 - 2 º TR = recrystallization temperature TR Adapted from Fig. 7.22, Callister 7e. The influence of annealing T on the tensile strength and ductility of a brass alloy. º Chapter 10 - 3 Iron-Carbon Phase Diagram Eutectoid cooling: cool g (0.76 wt% C) a (0.022 wt% C) + Fe3C (6.7 wt% C) T(°C) heat 1600 d 1400 L g g+L 1200 A L+Fe C (austenite) 1148°C 3 R S 1000 g g g+Fe C g g 3 800a B 727°C = Teutectoid R S (cementite) C 3 600 a+Fe C 3 Fe 400 0 1 2 3 4 5 6 6.7 0.76 4.30 (Fe) Co, wt% C Fe3C (cementite-hard) a (ferrite-soft) eutectoid Adapted from Fig. -

MSE3 Ch07 Precipitation
Chapter 7 Copyright © 2011, 2015 by Roland Stull. Meteorology for Scientists and Engineers, 3rd Ed. preCipitation Contents Hydrometeors are liquid and ice parti- cles that form in the atmosphere. Hydrom- Supersaturation and Water Availability 186 eter sizes range from small cloud droplets Supersaturation 186 7 and ice crystals to large hailstones. Precipitation Water Availability 186 occurs when hydrometeors are large and heavy Number and Size of Hydrometeors 187 enough to fall to the Earth’s surface. Virga occurs Nucleation of Liquid Droplets 188 when hydrometers fall from a cloud, but evaporate Cloud Condensation Nuclei (CCN) 188 before reaching the Earth’s surface. Curvature and Solute Effects 189 Precipitation particles are much larger than cloud Critical Radius 192 particles, as illustrated in Fig. 7.1. One “typical” Haze 192 Activated Nuclei 193 raindrop holds as much water as a million “typical” cloud droplets. How do such large precipitation Nucleation of Ice Crystals 194 Processes 194 particles form? Ice Nuclei 195 The microphysics of cloud- and precipitation- particle formation is affected by super-saturation, Liquid Droplet Growth by Diffusion 196 nucleation, diffusion, and collision. Ice Growth by Diffusion 198 Supersaturation indicates the amount of excess Ice Crystal Habits 198 water vapor available to form rain and snow. Growth Rates 200 The Wegener-Bergeron-Findeisen (WBF) Nucleation is the formation of new liquid or Process 201 solid hydrometeors as water vapor attaches to tiny dust particles carried in the air. These particles are Collision and Collection 202 Terminal Velocity of Hydrometeors 202 called cloud condensation nuclei or ice nuclei. Cloud Droplets 202 Diffusion is the somewhat random migration Rain Drops 203 of water-vapor molecules through the air toward ex- Hailstones 204 isting hydrometeors. -

On the Homogeneous and Heterogeneous Nucleation of Some Organic Compounds
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Helsingin yliopiston digitaalinen arkisto UNIVERSITY OF HELSINKI ON THE HOMOGENEOUS AND HETEROGENEOUS NUCLEATION OF SOME ORGANIC COMPOUNDS by Anca Ioana Hienola Academic dissertation To be presented, with the permission of the Faculty of Science of the University of Helsinki, for public criticism in auditorium D101 Gustaf H¨allstr¨ominkatu 2 , on June 4th, 2008, at 12 o'clock Helsinki 2008 \The most exciting phrase to hear in science, the one that heralds the most discoveries, is not Eureka! (I found it!) but 'That's funny... " Isaac Asimov UNIVERSITY OF HELSINKI Abstract On the homogeneous and heterogeneous nucleation of some organic compounds Anca Ioana Hienola The conversion of a metastable phase into a thermodynamically stable phase takes place via the formation of clusters. Clusters of different sizes are formed spontaneously within the metastable mother phase, but only those larger than a certain size, called the critical size, will end up growing into a new phase. There are two types of nucleation: homogeneous, where the clusters appear in a uniform phase, and heterogeneous, when pre-existing surfaces are available and clusters form on them. The nucleation of aerosol particles from gas-phase molecules is connected not only with inorganic compounds, but also with nonvolatile organic substances found in atmosphere. The question is which ones of the myriad of organic species have the right properties and are able to participate in nucleation phenomena. This thesis discusses both homogeneous and heterogeneous nucleation, having as theo- retical tool the classical nucleation theory (CNT) based on thermodynamics.