Introduction to the Physics of Nucleation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Colloidal Crystallization: Nucleation and Growth
Colloidal Crystallization: Nucleation and Growth Dave Weitz Harvard Urs Gasser Konstanz Andy Schofield Edinburgh Rebecca Christianson Harvard Peter Pusey Edinburgh Peter Schall Harvard Frans Spaepen Harvard Eric Weeks Emory John Crocker UPenn •Colloids as model systems – soft materials •Nucleation and growth of colloidal crystals •Defect growth in crystals •Binary Alloy Colloidal Crystals NSF, NASA http://www.deas.harvard.edu/projects/weitzlab EMU 6/7//04 Colloidal Crystals Colloids 1 nm - 10 µm solid particles in a solvent Ubiquitous ink, paint, milk, butter, mayonnaise, toothpaste, blood Suspensions can act like both liquid and solid Modify flow properties Control: Size, uniformity, interactions Colloidal Particles •Solid particles in fluid •Hard Spheres •Volume exclusion •Stability: •Short range repulsion •Sometimes a slight charge Colloid Particles are: •Big Slow •~ a ~ 1 micron • τ ~ a2/D ~ ms to sec •Can “see” them •Follow individual particle dynamics Model: Colloid Æ Atom Phase Behavior Colloids Atomic Liquids U U s s Hard Spheres Leonard-Jones Osmotic Pressure Attraction Centro-symmetric Centro-symmetric Thermalized: kBT Thermalized: kBT Gas Gas Liquid Density Liquid Solid Solid Phase behavior is similar Hard Sphere Phase Diagram Volume Fraction Controls Phase Behavior liquid - crystal liquid coexistence crystal 49% 54% 63% 74% φ maximum packing maximum packing φRCP≈0.63 φHCP=0.74 0 Increase φ => Decrease Temperature F = U - TS Entropy Drives Crystallization 0 Entropy => Free Volume F = U - TS Disordered: Ordered: •Higher configurational -

Entropy and the Tolman Parameter in Nucleation Theory
entropy Article Entropy and the Tolman Parameter in Nucleation Theory Jürn W. P. Schmelzer 1 , Alexander S. Abyzov 2 and Vladimir G. Baidakov 3,* 1 Institute of Physics, University of Rostock, Albert-Einstein-Strasse 23-25, 18059 Rostock, Germany 2 National Science Center Kharkov Institute of Physics and Technology, 61108 Kharkov, Ukraine 3 Institute of Thermal Physics, Ural Branch of the Russian Academy of Sciences, Amundsen Street 107a, 620016 Yekaterinburg, Russia * Correspondence: [email protected]; Tel.: +49-381-498-6889; Fax: +49-381-498-6882 Received: 23 May 2019; Accepted: 5 July 2019; Published: 9 July 2019 Abstract: Thermodynamic aspects of the theory of nucleation are commonly considered employing Gibbs’ theory of interfacial phenomena and its generalizations. Utilizing Gibbs’ theory, the bulk parameters of the critical clusters governing nucleation can be uniquely determined for any metastable state of the ambient phase. As a rule, they turn out in such treatment to be widely similar to the properties of the newly-evolving macroscopic phases. Consequently, the major tool to resolve problems concerning the accuracy of theoretical predictions of nucleation rates and related characteristics of the nucleation process consists of an approach with the introduction of the size or curvature dependence of the surface tension. In the description of crystallization, this quantity has been expressed frequently via changes of entropy (or enthalpy) in crystallization, i.e., via the latent heat of melting or crystallization. Such a correlation between the capillarity phenomena and entropy changes was originally advanced by Stefan considering condensation and evaporation. It is known in the application to crystal nucleation as the Skapski–Turnbull relation. -

Observation of Elliptically Polarized Light from Total Internal Reflection in Bubbles
Observation of elliptically polarized light from total internal reflection in bubbles Item Type Article Authors Miller, Sawyer; Ding, Yitian; Jiang, Linan; Tu, Xingzhou; Pau, Stanley Citation Miller, S., Ding, Y., Jiang, L. et al. Observation of elliptically polarized light from total internal reflection in bubbles. Sci Rep 10, 8725 (2020). https://doi.org/10.1038/s41598-020-65410-5 DOI 10.1038/s41598-020-65410-5 Publisher NATURE PUBLISHING GROUP Journal SCIENTIFIC REPORTS Rights Copyright © The Author(s) 2020. Open Access This article is licensed under a Creative Commons Attribution 4.0 International License. Download date 29/09/2021 02:08:57 Item License https://creativecommons.org/licenses/by/4.0/ Version Final published version Link to Item http://hdl.handle.net/10150/641865 www.nature.com/scientificreports OPEN Observation of elliptically polarized light from total internal refection in bubbles Sawyer Miller1,2 ✉ , Yitian Ding1,2, Linan Jiang1, Xingzhou Tu1 & Stanley Pau1 ✉ Bubbles are ubiquitous in the natural environment, where diferent substances and phases of the same substance forms globules due to diferences in pressure and surface tension. Total internal refection occurs at the interface of a bubble, where light travels from the higher refractive index material outside a bubble to the lower index material inside a bubble at appropriate angles of incidence, which can lead to a phase shift in the refected light. Linearly polarized skylight can be converted to elliptically polarized light with efciency up to 53% by single scattering from the water-air interface. Total internal refection from air bubble in water is one of the few sources of elliptical polarization in the natural world. -

Thermodynamics and Kinetics of Nucleation in Binary Solutions
1 Thermodynamics and kinetics of nucleation in binary solutions Nikolay V. Alekseechkin Akhiezer Institute for Theoretical Physics, National Science Centre “Kharkov Institute of Physics and Technology”, Akademicheskaya Street 1, Kharkov 61108, Ukraine Email: [email protected] Keywords: Binary solution; phase equilibrium; binary nucleation; diffusion growth; surface layer. A new approach that is a combination of classical thermodynamics and macroscopic kinetics is offered for studying the nucleation kinetics in condensed binary solutions. The theory covers the separation of liquid and solid solutions proceeding along the nucleation mechanism, as well as liquid-solid transformations, e.g., the crystallization of molten alloys. The cases of nucleation of both unary and binary precipitates are considered. Equations of equilibrium for a critical nucleus are derived and then employed in the macroscopic equations of nucleus growth; the steady state nucleation rate is calculated with the use of these equations. The present approach can be applied to the general case of non-ideal solution; the calculations are performed on the model of regular solution within the classical nucleation theory (CNT) approximation implying the bulk properties of a nucleus and constant surface tension. The way of extending the theory beyond the CNT approximation is shown in the framework of the finite- thickness layer method. From equations of equilibrium of a surface layer with coexisting bulk phases, equations for adsorption and the dependences of surface tension on temperature, radius, and composition are derived. Surface effects on the thermodynamics and kinetics of nucleation are discussed. 1. Introduction Binary nucleation covers a wide class of processes of phase transformations which can be divided into three groups: (i) gas-liquid (or solid) transformations, (ii) liquid-gas transformations, and (iii) transformations within a condensed state. -

Heterogeneous Nucleation of Water Vapor on Different Types of Black Carbon Particles
https://doi.org/10.5194/acp-2020-202 Preprint. Discussion started: 21 April 2020 c Author(s) 2020. CC BY 4.0 License. Heterogeneous nucleation of water vapor on different types of black carbon particles Ari Laaksonen1,2, Jussi Malila3, Athanasios Nenes4,5 5 1Finnish Meteorological Institute, 00101 Helsinki, Finland. 2Department of Applied Physics, University of Eastern Finland, 70211 Kuopio, Finland. 3Nano and Molecular Systems Research Unit, 90014 University of Oulu, Finland. 4Laboratory of Atmospheric Processes and their Impacts, School of Architecture, Civil & Environmental Engineering, École 10 Polytechnique Federale de Lausanne, 1015, Lausanne, Switzerland 5 Institute of Chemical Engineering Sciences, Foundation for Research and Technology Hellas (FORTH/ICE-HT), 26504, Patras, Greece Correspondence to: Ari Laaksonen ([email protected]) 15 Abstract: Heterogeneous nucleation of water vapor on insoluble particles affects cloud formation, precipitation, the hydrological cycle and climate. Despite its importance, heterogeneous nucleation remains a poorly understood phenomenon that relies heavily on empirical information for its quantitative description. Here, we examine heterogeneous nucleation of water vapor on and cloud drop activation of different types of soots, both pure black carbon particles, and black carbon particles mixed with secondary organic matter. We show that the recently developed adsorption nucleation theory 20 quantitatively predicts the nucleation of water and droplet formation upon particles of the various soot types. A surprising consequence of this new understanding is that, with sufficient adsorption site density, soot particles can activate into cloud droplets – even when completely lacking any soluble material. 1 https://doi.org/10.5194/acp-2020-202 Preprint. Discussion started: 21 April 2020 c Author(s) 2020. -

A Monte Carlo Ray Tracing Study of Polarized Light Propagation in Liquid Foams
ARTICLE IN PRESS Journal of Quantitative Spectroscopy & Radiative Transfer 104 (2007) 277–287 www.elsevier.com/locate/jqsrt A Monte Carlo ray tracing study of polarized light propagation in liquid foams J.N. Swamya,Ã, Czarena Crofchecka, M. Pinar Mengu¨c- b,ÃÃ aDepartment of Biosystems and Agricultural Engineering, 128 C E Barnhart Building, University of Kentucky, Lexington, KY 40546, USA bDepartment of Mechanical Engineering, 269 Ralph G. Anderson Building, University of Kentucky, Lexington, KY 40506, USA Received 10 July 2006; accepted 28 July 2006 Abstract A Monte Carlo ray tracing scheme is used to investigate the propagation of an incident collimated beam of polarized light in liquid foams. Cellular structures like foam are expected to change the polarization characteristics due to multiple scattering events, where such changes can be used to monitor foam dynamics. A statistical model utilizing some of the recent developments in foam physics is coupled with a vector Monte Carlo scheme to compute the depolarization ratios via Stokes–Mueller formalism. For the simulations, the incident Stokes vector corresponding to horizontal linear polarization and right circular polarization are considered. It is observed that bubble size and the polydispersity parameter have a significant effect on the depolarization ratios. This is partially owing to the number of total internal reflection events in the Plateau borders. The results are discussed in terms of applicability of polarized light as a diagnostic tool for monitoring foams. r 2006 Elsevier Ltd. All rights reserved. Keywords: Polarized light scattering; Liquid foams; Bubble size; Polydispersity; Foam characterization; Foam diagnostics; Cellular structures 1. Introduction Liquid foams are random packing of bubbles in a small amount of immiscible liquid [1] and can be found in a wide variety of applications. -

The Synergistic Effect of Focused Ultrasound and Biophotonics to Overcome the Barrier of Light Transmittance in Biological Tissue
Photodiagnosis and Photodynamic Therapy 33 (2021) 102173 Contents lists available at ScienceDirect Photodiagnosis and Photodynamic Therapy journal homepage: www.elsevier.com/locate/pdpdt The synergistic effect of focused ultrasound and biophotonics to overcome the barrier of light transmittance in biological tissue Jaehyuk Kim a,b, Jaewoo Shin c, Chanho Kong c, Sung-Ho Lee a, Won Seok Chang c, Seung Hee Han a,d,* a Molecular Imaging, Princess Margaret Cancer Centre, Toronto, ON, Canada b Health and Medical Equipment, Samsung Electronics Co. Ltd., Suwon, Republic of Korea c Department of Neurosurgery, Brain Research Institute, Yonsei University College of Medicine, Seoul, Republic of Korea d Department of Medical Biophysics, University of Toronto, Toronto, ON, Canada ARTICLE INFO ABSTRACT Keywords: Optical technology is a tool to diagnose and treat human diseases. Shallow penetration depth caused by the high Light transmission enhancement optical scattering nature of biological tissues is a significantobstacle to utilizing light in the biomedical field. In Focused Ultrasound this paper, light transmission enhancement in the rat brain induced by focused ultrasound (FUS) was observed Air bubble and the cause of observed enhancement was analyzed. Both air bubbles and mechanical deformation generated Mechanical deformation by FUS were cited as the cause. The Monte Carlo simulation was performed to investigate effects on transmission Rat brain by air bubbles and finiteelement method was also used to describe mechanical deformation induced by motions of acoustic particles. As a result, it was found that the mechanical deformation was more suitable to describe the transmission change according to the FUS pulse observed in the experiment. 1. -

Ocean Storage
277 6 Ocean storage Coordinating Lead Authors Ken Caldeira (United States), Makoto Akai (Japan) Lead Authors Peter Brewer (United States), Baixin Chen (China), Peter Haugan (Norway), Toru Iwama (Japan), Paul Johnston (United Kingdom), Haroon Kheshgi (United States), Qingquan Li (China), Takashi Ohsumi (Japan), Hans Pörtner (Germany), Chris Sabine (United States), Yoshihisa Shirayama (Japan), Jolyon Thomson (United Kingdom) Contributing Authors Jim Barry (United States), Lara Hansen (United States) Review Editors Brad De Young (Canada), Fortunat Joos (Switzerland) 278 IPCC Special Report on Carbon dioxide Capture and Storage Contents EXECUTIVE SUMMARY 279 6.7 Environmental impacts, risks, and risk management 298 6.1 Introduction and background 279 6.7.1 Introduction to biological impacts and risk 298 6.1.1 Intentional storage of CO2 in the ocean 279 6.7.2 Physiological effects of CO2 301 6.1.2 Relevant background in physical and chemical 6.7.3 From physiological mechanisms to ecosystems 305 oceanography 281 6.7.4 Biological consequences for water column release scenarios 306 6.2 Approaches to release CO2 into the ocean 282 6.7.5 Biological consequences associated with CO2 6.2.1 Approaches to releasing CO2 that has been captured, lakes 307 compressed, and transported into the ocean 282 6.7.6 Contaminants in CO2 streams 307 6.2.2 CO2 storage by dissolution of carbonate minerals 290 6.7.7 Risk management 307 6.2.3 Other ocean storage approaches 291 6.7.8 Social aspects; public and stakeholder perception 307 6.3 Capacity and fractions retained -

Determination of Supercooling Degree, Nucleation and Growth Rates, and Particle Size for Ice Slurry Crystallization in Vacuum
crystals Article Determination of Supercooling Degree, Nucleation and Growth Rates, and Particle Size for Ice Slurry Crystallization in Vacuum Xi Liu, Kunyu Zhuang, Shi Lin, Zheng Zhang and Xuelai Li * School of Chemical Engineering, Fuzhou University, Fuzhou 350116, China; [email protected] (X.L.); [email protected] (K.Z.); [email protected] (S.L.); [email protected] (Z.Z.) * Correspondence: [email protected] Academic Editors: Helmut Cölfen and Mei Pan Received: 7 April 2017; Accepted: 29 April 2017; Published: 5 May 2017 Abstract: Understanding the crystallization behavior of ice slurry under vacuum condition is important to the wide application of the vacuum method. In this study, we first measured the supercooling degree of the initiation of ice slurry formation under different stirring rates, cooling rates and ethylene glycol concentrations. Results indicate that the supercooling crystallization pressure difference increases with increasing cooling rate, while it decreases with increasing ethylene glycol concentration. The stirring rate has little influence on supercooling crystallization pressure difference. Second, the crystallization kinetics of ice crystals was conducted through batch cooling crystallization experiments based on the population balance equation. The equations of nucleation rate and growth rate were established in terms of power law kinetic expressions. Meanwhile, the influences of suspension density, stirring rate and supercooling degree on the process of nucleation and growth were studied. Third, the morphology of ice crystals in ice slurry was obtained using a microscopic observation system. It is found that the effect of stirring rate on ice crystal size is very small and the addition of ethylene glycoleffectively inhibits the growth of ice crystals. -

Bubble-Mania the Bubbling Clues to Magma Viscosity and Eruptions
Earthlearningidea - http://www.earthlearningidea.com/ Bubble-mania The bubbling clues to magma viscosity and eruptions Pour a viscous liquid (eg. honey, syrup) into Honey and a one transparent container and a pale-coloured soft drink – ready for soft drink (eg. ginger beer) or just coloured bubble-mania. water into another. Put both of these onto a plastic tray or table. Ask your pupils to use a drinking straw to blow bubbles into the soft Apparatus photo: drink, then ask them to ‘use the same blow’ to Chris King blow bubbles in the more viscous liquid. When nothing happens, ask them to blow harder until the liquid ‘erupts’. Ask: • How were the ‘eruptions’ different? • How were the bubbles different? • What caused the differences? • Some volcanoes have magmas that are Magma fountain ‘runny’ (like the soft drink) and some have within the crater much more viscous magmas (like the other of Volcan liquid) – how might these volcanoes erupt Villarrica, Pucón, differently? Chile. • Which sort of eruption would you most like to This file is see – one with low viscosity (runny) magma, licensed by Jonathan Lewis like the soft drink, or one with high viscosity under the Creative (thick) magma like the viscous liquid? Commons Attribution-Share Alike 2.0 Generic license. ……………………………………………………………………………………………………………………………… The back up Title: Bubble-mania • How were the ‘eruptions’ different? It was easy to blow bubbles into the soft drink Subtitle: The bubbling clues to magma viscosity and it ‘fizzed’ a bit, but the bubbles soon and eruptions disappeared; it was much harder to blow bubbles into the viscous liquid and they grew Topic: A simple test of the viscosity of two similar- large and sometimes burst out of the container looking liquids, linked to volcanic eruption style. -
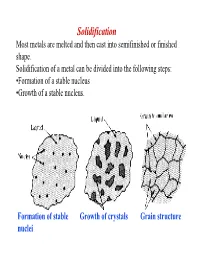
Solidification Most Metals Are Melted and Then Cast Into Semifinished Or Finished Shape
Solidification Most metals are melted and then cast into semifinished or finished shape. Solidification of a metal can be divided into the following steps: •Formation of a stable nucleus •Growth of a stable nucleus. Formation of stable Growth of crystals Grain structure nuclei Polycrystalline Metals In most cases, solidification begins from multiple sites, each of which can produce a different orientation The result is a “polycrystalline” material consisting of many small crystals of “grains” Each grain has the same crystal lattice, but the lattices are misoriented from grain to grain Driving force: solidification ⇒ For the reaction to proceed to the right ∆G AL AS V must be negative. • Writing the free energies of the solid and liquid as: S S S GV = H - TS L L L GV = H - TS ∴∴∴ ∆GV = ∆H - T∆S • At equilibrium, i.e. Tmelt , then the ∆GV = 0 , so we can estimate the melting entropy as: ∆S = ∆H/Tmelt where -∆H is the latent heat (enthalpy) of melting. • Ignore the difference in specific heat between solid and liquid, and we estimate the free energy difference as: T ∆H × ∆T ∆GV ≅ ∆H − ∆H = TMelt TMelt NUCLEATION The two main mechanisms by which nucleation of a solid particles in liquid metal occurs are homogeneous and heterogeneous nucleation. Homogeneous Nucleation Homogeneous nucleation occurs when there are no special objects inside a phase which can cause nucleation. For instance when a pure liquid metal is slowly cooled below its equilibrium freezing temperature to a sufficient degree numerous homogeneous nuclei are created by slow-moving atoms bonding together in a crystalline form. -

The Role of Latent Heat in Heterogeneous Ice Nucleation
EGU21-16164, updated on 02 Oct 2021 https://doi.org/10.5194/egusphere-egu21-16164 EGU General Assembly 2021 © Author(s) 2021. This work is distributed under the Creative Commons Attribution 4.0 License. The role of latent heat in heterogeneous ice nucleation Olli Pakarinen, Cintia Pulido Lamas, Golnaz Roudsari, Bernhard Reischl, and Hanna Vehkamäki University of Helsinki, INAR/Physics, University of Helsinki, Finland ([email protected]) Understanding the way in which ice forms is of great importance to many fields of science. Pure water droplets in the atmosphere can remain in the liquid phase to nearly -40º C. Crystallization of ice in the atmosphere therefore typically occurs in the presence of ice nucleating particles (INPs), such as mineral dust or organic particles, which trigger heterogeneous ice nucleation at clearly higher temperatures. The growth of ice is accompanied by a significant release of latent heat of fusion, which causes supercooled liquid droplets to freeze in two stages [Pruppacher and Klett, 1997]. We are studying these topics by utilizing the monatomic water model [Molinero and Moore, 2009] for unbiased molecular dynamics (MD) simulations, where different surfaces immersed in water are cooled below the melting point over tens of nanoseconds of simulation time and crystallization is followed. With a combination of finite difference calculations and novel moving-thermostat molecular dynamics simulations we show that the release of latent heat from ice growth has a noticeable effect on both the ice growth rate and the initial structure of the forming ice. However, latent heat is found not to be as critically important in controlling immersion nucleation as it is in vapor-to- liquid nucleation [Tanaka et al.2017].