Fereshteh Samadani Liquid Chemistry of Hassium Tetroxide Using
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Physical Review Journals Catalog 2021
2021 PHYSICAL REVIEW JOURNALS CATALOG PUBLISHED BY THE AMERICAN PHYSICAL SOCIETY Physical Review Journals 2021 1 © 2020 American Physical Society 2 Physical Review Journals 2021 Table of Contents Founded in 1899, the American Physical Society (APS) strives to advance and diffuse the knowledge of physics. In support of this objective, APS publishes primary research and review journals, five of which are open access. Physical Review Letters..............................................................................................................2 Physical Review X .......................................................................................................................3 PRX Quantum .............................................................................................................................4 Reviews of Modern Physics ......................................................................................................5 Physical Review A .......................................................................................................................6 Physical Review B ......................................................................................................................7 Physical Review C.......................................................................................................................8 Physical Review D ......................................................................................................................9 Physical Review E ................................................................................................................... -

The Separation and Determination of Osmium and Ruthenium
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1969 The epS aration and Determination of Osmium and Ruthenium. Harry Edward Moseley Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Moseley, Harry Edward, "The eS paration and Determination of Osmium and Ruthenium." (1969). LSU Historical Dissertations and Theses. 1559. https://digitalcommons.lsu.edu/gradschool_disstheses/1559 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. ThU dissertation has been 69-17,123 microfilmsd exactly as received MOSELEY, Harry Edward, 1929- THE SEPARATION AND DETERMINATION OF OSMIUM AND RUTHENIUM. Louisiana State University and Agricultural and Mechanical College, PhJ>., 1969 Chemistry, analytical University Microfilms, Inc., Ann Arbor, Michigan THE SEPARATION AND DETERMINATION OF OSMIUM AND RUTHENIUM A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The Department of Chemistry Harry Edward Moseley B.S., Lpuisiana State University, 1951 M.S., Louisiana State University, 1952 J anuary, 1969 ACKNOWLEDGMENTS Thanks are due to Dr. Eugene W. Berg under whose direction this work was performed, to Dr. A. D. Shendrikar for his help in the tracer studies, and to Mr. J. H. R. Streiffer for his help in writing the com puter program. -

Citation Statistics from 110 Years of Physical Review
Citation Statistics from 110 Years of Physical Review Publicly available data reveal long-term systematic features about citation statistics and how papers are referenced. The data also tell fascinating citation histories of individual articles. Sidney Redner he first article published in the Physical Review was Treceived in 1893; the journal’s first volume included 6 issues and 24 articles. In the 20th century, the Phys- ical Review branched into topical sections and spawned new journals (see figure 1). Today, all arti- cles in the Physical Review family of journals (PR) are available online and, as a useful byproduct, all cita- tions in PR articles are electronically available. The citation data provide a treasure trove of quantitative information. As individuals who write sci- entific papers, most of us are keenly interested in how often our own work is cited. As dispassionate observers, we Figure 1. The Physical Review can use the citation data to identify influential research, was the first member of a family new trends in research, unanticipated connections across of journals that now includes two series of Physical fields, and downturns in subfields that are exhausted. A Review, the topical journals Physical Review A–E, certain pleasure can also be gleaned from the data when Physical Review Letters (PRL), Reviews of Modern Physics, they reveal the idiosyncratic features in the citation his- and Physical Review Special Topics: Accelerators and tories of individual articles. Beams. The first issue of Physical Review bears a publica- The investigation of citation statistics has a long his- tion date a year later than the receipt of its first article. -

PHYSICAL REVIEW a Covering Atomic, Molecular, and Optical Physics and Quantum Information
PHYSICAL REVIEW A covering atomic, molecular, and optical physics and quantum information EDITORIAL BOARD Term ending 31 December 2020 M. HALL Quantum Optics and Information, Foundations of Quantum Mechanics, Open Quantum Systems Editor P. KOK Quantum Optics and Quantum Information American Physical Society JAN MICHAEL ROST B. KRAUS Quantum Information Theory (Max Planck Institute for the Physics F. ROBICHEAUX Time-dependent Atomic Phenomena, President PHILIP H. BUCKSBAUM Speaker of the Council ANDREA J. LIU of Complex Systems) Highly excited Atoms, and Ultracold Plasmas Chief Executive Officer KATE P. KIRBY G. STEINMEYER Ultrafast Lasers and Phenomena, Nonlinear Optics Managing Editor Editor in Chief MICHAEL THOENNESSEN J. THYWISSEN Ultracold Atoms THOMAS PATTARD Publisher MATTHEW SALTER M. XIAO Quantum Optics and Atomic Coherence Chief Information Officer MARK D. DOYLE (APS Editorial Office) Term ending 31 December 2021 Chief Financial Officer JANE HOPKINS GOULD Associate Editors A. J. DALEY Cold Quantum Gases and Quantum Optics Deputy Executive Officer and Chief Operating Officer JAMES TAYLOR UGO ANCARANI G. JOHANSSON Quantum Physics and Quantum Information (Universite´ de Lorraine) Chief Government Affairs with Superconducting Circuits Officer FRANCIS SLAKEY ERIKA ANDERSSON G. KLIMCHITSKAYA Fluctuation Phenomena, Casimir Forces, (Heriot-Watt University) One Physics Ellipse Atom-Surface Interactions College Park, MD 20740-3844 DIRK JAN BUKMAN L. B. MADSEN Attosecond Physics, and Atoms and (APS Editorial Office) Molecules in Strong Fields KATIUSCIA CASSEMIRO S. PASCAZIO Cold Atoms, Entanglement, and Quantum Information (APS Editorial Office) S. G. PORSEV Atomic Theory and Structure APS Editorial Office FRANCO DALFOVO H. PU Theory of Quantum Gases Directors (Universita` di Trento) B. M. TERHAL Quantum Information Journal Operations CHRISTINE M. -

Evolution of Intrinsic Nuclear Structure in Medium Mass Even-Even Xenon Isotopes from a Microscopic Perspective*
Chinese Physics C Vol. 44, No. 7 (2020) 074108 Evolution of intrinsic nuclear structure in medium mass even-even Xenon isotopes from a microscopic perspective* Surbhi Gupta1 Ridham Bakshi1 Suram Singh2 Arun Bharti1;1) G. H. Bhat3 J. A. Sheikh4 1Department of Physics, University of Jammu, Jammu- 180006, India 2Department of Physics and Astronomical Sciences, Central University of Jammu, Samba- 181143, India 3Department of Physics, SP College, Cluster University Srinagar- 190001, India 4Cluster University Srinagar - Jammu and Kashmir 190001, India Abstract: In this study, the multi-quasiparticle triaxial projected shell model (TPSM) is applied to investigate γ-vi- brational bands in transitional nuclei of 118−128Xe. We report that each triaxial intrinsic state has a γ-band built on it. The TPSM approach is evaluated by the comparison of TPSM results with available experimental data, which shows a satisfactory agreement. The energy ratios, B(E2) transition rates, and signature splitting of the γ-vibrational band are calculated. Keywords: triaxial projected shell model, triaxiality, yrast spectra, band diagram, back-bending, staggering, reduced transition probabilities DOI: 10.1088/1674-1137/44/7/074108 1 Introduction In the past few decades, the use of improved and sophisticated experimental techniques has made it pos- sible to provide sufficient data to describe the structure of The static and dynamic properties of a nucleus pre- nuclei in various mass regions of the nuclear chart. In dominantly dictate its shape or structure, and these prop- particular, the transitional nuclei around mass A ∼ 130 erties depend on the interactions among its constituents, have numerous interesting features, such as odd-even i.e, protons and neutrons. -

Writing Physics Papers
Physics 2151W Lab Manual | Page 45 WID Handbook for Intermediate Laboratory - Physics 2151W Writing Physics Papers Dr. Igor Strakovsky Department of Physics, GWU Publish or Perish - Presentation of Scientific Results Intermediate Laboratory – Physics 2151W is focused on significantly improving the students' writing skills with respect to producing scientific papers, to do peer reviews, and presentations at the Physics Department Mini-Workshop. Third Edition, 2013 Physics 2151W Lab Manual | Page 46 OUTLINE Why are we Writing Papers? What Physics Journals are there? Structure of a Physics Article. Style of Technical Papers. Hints for Effective Writing. Submit and Fight. Why are We Writing Papers? To communicate our original, interesting, and useful research. To let others know what we are working on (and that we are working at all.) To organize our thoughts. To formulate our research in a comprehensible way. To secure further funding. To further our careers. To make our publication lists look more impressive. To make our Citation Index very impressive. To have fun? Because we believe someone is going to read it!!! Physics 2151W Lab Manual | Page 47 What Physics Journals are there? Hard Science Journals Physical Review Series: Physical Review A Physical Review E http://pra.aps.org/ http://pre.aps.org/ Atomic, Molecular, and Optical physics. Stat, Non-Linear, & Soft Material Phys. Physical Review B Physical Review Letters http://prb.aps.org/ http://prl.aps.org/ Condensed matter and Materials physics. Moving physics forward. Physical Review C Review of Modern Physics http://prc.aps.org/ http://rmp.aps.org/ Nuclear physics. Reviews in all areas. Physical Review D http://prd.aps.org/ Particles, Fields, Gravitation, and Cosmology. -

Quest for Superheavy Nuclei Began in the 1940S with the Syn Time It Takes for Half of the Sample to Decay
FEATURES Quest for superheavy nuclei 2 P.H. Heenen l and W Nazarewicz -4 IService de Physique Nucleaire Theorique, U.L.B.-C.P.229, B-1050 Brussels, Belgium 2Department ofPhysics, University ofTennessee, Knoxville, Tennessee 37996 3Physics Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831 4Institute ofTheoretical Physics, University ofWarsaw, ul. Ho\.za 69, PL-OO-681 Warsaw, Poland he discovery of new superheavy nuclei has brought much The superheavy elements mark the limit of nuclear mass and T excitement to the atomic and nuclear physics communities. charge; they inhabit the upper right corner of the nuclear land Hopes of finding regions of long-lived superheavy nuclei, pre scape, but the borderlines of their territory are unknown. The dicted in the early 1960s, have reemerged. Why is this search so stability ofthe superheavy elements has been a longstanding fun important and what newknowledge can it bring? damental question in nuclear science. How can they survive the Not every combination ofneutrons and protons makes a sta huge electrostatic repulsion? What are their properties? How ble nucleus. Our Earth is home to 81 stable elements, including large is the region of superheavy elements? We do not know yet slightly fewer than 300 stable nuclei. Other nuclei found in all the answers to these questions. This short article presents the nature, although bound to the emission ofprotons and neutrons, current status ofresearch in this field. are radioactive. That is, they eventually capture or emit electrons and positrons, alpha particles, or undergo spontaneous fission. Historical Background Each unstable isotope is characterized by its half-life (T1/2) - the The quest for superheavy nuclei began in the 1940s with the syn time it takes for half of the sample to decay. -

Masthead (Print)
PHYSICAL REVIEW A ATOMIC, MOLECULAR, AND OPTICAL PHYSICS The American Physical Society Editor EDITORIAL BOARD One Physics Ellipse College Park, MD 20740-3844 GORDON W. F. DRAKE Term ending 31 December 2008 (University of Windsor) President ARTHUR BIENENSTOCK President-Elect CHERRY A. MURRAY J. S. COHEN Theoretical Physics Managing Editor Vice-President CURTIS G. CALLAN M. FLEISCHHAUER Quantum Optics MARGARET MALLOY Editor-in-Chief GENE D. SPROUSE D. J. GAUTHIER Quantum Optics Treasurer (APS Editorial Office) P. L. GOULD Atomic and Molecular Physics and Publisher JOSEPH W. SERENE Executive Officer JUDY R. FRANZ Associate Editors B. C. SANDERS Quantum Information G. V. SHLYAPNIKOV Matter Waves DIRK JAN BUKMAN L. VIOLA Quantum Information (APS Ediorial Office) LEE A. COLLINS APS Editorial Office Term ending 31 December 2009 (Los Alamos National Laboratory) 1 Research Road Ridge, NY 11961-2701 FRANCO DALFOVO A. D. BANDRAUK Short Laser Pulses (Universita` di Trento) M. J. HOLLAND Quantum Gases, Quantum Phone: (631) 591-4010 Fax: (631) 591-4141 JULIO GEA-BANACLOCHE Optics Email: [email protected] (University of Arkansas) E. LINDROTH Atomic Theory Web: http://publish.aps.org/ MARK HILLERY P. J. MOHR Atomic Theory Submissions: [email protected] or (Hunter College) C. J. PETHICK Matter Waves http://authors.aps.org/ESUB/ KEITH B. MACADAM J. ULLRICH Atomic and Molecular Structure (University of Kentucky) and Interactions FRANK NARDUCCI K. WODKIEWICZ´ Quantum Information, Quantum (Naval Air Systems Command) Optics Member Subscriptions: MARK SAFFMAN APS Membership Department (University of Wisconsin) Term ending 31 December 2010 One Physics Ellipse College Park, MD 20740-3844 Senior Assistant Editor I. BIALYNICKI-BIRULA Theoretical Physics Phone: (301) 209-3280 RUN Quantum Theory MARGARET FOSTER T. -

22.05 Reactor Physics – Part One Course Introduction
22.05 Reactor Physics – Part One Course Introduction 1. Instructor: John A. Bernard 2. Organization: Homework (20%) Four Exams (20% each; lowest grade is dropped) Final Exam (3.0 hours) (20%) 3. Text: The text book for this course is: Introduction to Nuclear Engineering, 3rd Edition, by John Lamarsh. This covers basic reactor physics as part of a complete survey of nuclear engineering. Readings may also be assigned from certain of the books listed below: Nuclear Reactor Analysis by A. F. Henry Introduction to Nuclear Power by G. Hewitt and J. Collier Fundamentals of Nuclear Science and Engineering by J. Shultis and R. Faw Atoms, Radiation, and Radiation Protection by J. Turner Nuclear Criticality Safety by R. Kneif Radiation Detection and Measurement by G. Knoll 4. Course Objective: To quote the late Professor Allan Henry: “The central problem of reactor physics can be stated quite simply. It is to compute, for any time t, the characteristics of the free-neutron population throughout an extended region of space containing an arbitrary, but known, mixture of materials. Specifically we wish to know the number of neutrons in any infinitesimal volume dV that have kinetic energies between E and E + ∆E and are traveling in directions within an infinitesimal angle of a fixed direction specified by the unit vector Ω. If this number is known, we can use the basic data obtained experimentally and theoretically from low-energy neutron physics to predict the rates at which all possible nuclear reactions, including fission, will take place throughout the region. Thus we can 1 predict how much nuclear power will be generated at any given time at any location in the region.” There are several reasons for needing this information: Physical understanding of reactor safety so that both design and operation is done intelligently. -
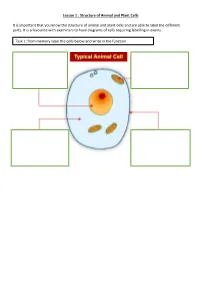
Lesson 1 : Structure of Animal and Plant Cells
Lesson 1 : Structure of Animal and Plant Cells It is important that you know the structure of animal and plant cells and are able to label the different parts. It is a favourite with examiners to have diagrams of cells requiring labelling in exams. Task 1: from memory label the cells below and write in the function Check your answers: There are many similarities and differences between animal and plant cells. Make sure you know these. Similarities Differences 1. Have a nucleus 1. Plant cells have a cellulose cell wall 2. Have a cytoplasm 2. Plant cells have a vacuole containing cell sap 3. Have a cell 3. Plant cells have chloroplast membrane 4. Contain 4. Many plant cells have a box-like shape whilst animal cell shape varies mitochondria 5. Plant cells have the nucleus to the side of the cell, animal cells have a nucleus in 5. Contain ribosomes the middle Task 2: Complete the sentences by filling in the gaps. Both plant and animal cells contain a nucleus. This holds genetic information. Both animal and plant cells have a cell membrane. This controls what enters and leaves the cell. Only a plant cell contains chloroplasts. This is where photosynthesis happens. Both cells contain mitochondria. This is where respiration occurs. Check your answers: Both plant and animal cells contain a nucleus. This holds genetic information. Both animal and plant cells have a cell membrane. This controls what enters and leaves the cell. Only a plant cell contains chloroplasts. This is where photosynthesis happens. Both cells contain mitochondria. This is where respiration occurs. -

Jahresbericht 2007
IKMz 2008- 1 ISSN 0932-7622 INSTITUT FÜR KERNCHEMIE UNIVERSITÄT MAINZ JAHRESBERICHT 2007 August 2008 FRITZ-STRASSMANN-WEG 2, D-55128 MAINZ, TELEFON (06131) 3925879, TELEFAX (06131) 3925253 Kernchemie im Internet Das Institut für Kernchemie der Universität Mainz ist mit einer Homepage im World Wide Web vertreten. Unter der Adresse http://www.kernchemie.uni-mainz.de finden Sie aktuelle Informationen zum Institut, seinen Mitarbeitern, den Lehrveranstaltungen und den Forschungsaktivitäten. Die in diesem Bericht vorgelegten Ergebnisse stammen zum Teil aus noch nicht abgeschlossenen Arbeiten und sind daher als vorläufige Mitteilung zu bewerten. INSTITUT FÜR KERNCHEMIE UNIVERSITÄT MAINZ JAHRESBERICHT 2007 Herausgeber: Frank Rösch Mainz, im August 2008 Vorwort Der vorliegende Jahresbericht 2007 gibt einen Überblick über die wissenschaftlichen Aktivitäten des Instituts für Kernchemie. Er legt damit gleichzeitig all denen, die uns in ideeller und finanzieller Weise gefördert haben, Rechenschaft ab über die Verwendung nicht unerheblicher öffentlicher Mittel. Außerdem beschreibt er den Status der technischen Einrichtungen des Instituts und technische Neu- entwicklungen. Schließlich stellt er die Ergebnisse des Instituts in Form von Publikationen, Konferenzbeiträgen, Dissertationen, Diplom- und Staatsexamensarbeiten sowie die Beiträge seiner Hochschullehrer in der Lehre und Weiterbildung dar. Der wissenschaftliche Bericht umfasst die drei Forschungsschwerpunkte: • Kernchemie im Sinne grundlegender Fragestellungen, • Radiopharmazeutische Chemie und Anwendung radiochemischer Methoden mit medizinischer Zielsetzung, • Hochempfindliche und –selektive Analytik für umweltrelevante, technische und biologische Probleme. Im August 2007 hat der Präsident unserer Universität, Herr Prof. Dr. Krausch, die Laufzeit des Forschungsreaktors TRIGA Mainz bis zunächst 2020 verlängert. Die Carl-Zeiss-Stiftung hat 2007 die Einrichtung einer Juniorprofessur für das Thema „Ultrakalte Neutronen“ am Institut für Kernchemie ab 2008 ermöglicht. -

Physics of Superheavy Elements Kouichi Hagino
Frontiers in Science II 2013.11.6 Physics of superheavy elements Kouichi Hagino Nuclear Theory Group, Department of Physics, Tohoku University What is nuclear physics? What are superheavy elements? How to create superheavy elements? What are chemical properties of superheavy elements? Introduction: atoms and atomic nuclei What would you see if you magnified the dog? ~ 50 cm Introduction: atoms and atomic nuclei cells ~ 50 cm ~ m = 10-6 m Introduction: atoms and atomic nuclei DNA cells -8 ~ 50 cm ~ m = 10-6 m ~ 10 m atom All things are made of atoms. ~ 10-10 m All things are made of atoms. • Thales, Democritus (ancient Greek) • Dalton (chemist, 19th century) • Boltzmann(19th century) • Einstein (1905) ~ 10-10 m STM image (surface physics group, Tohoku university) Introduction: atoms and atomic nuclei DNA cells -8 ~ 50 cm ~ 10 m atom atomic nucleus ~ 10-15 m ~ 10-10 m proton (+e) neutron (no charge) electron cloud (-e) Neutral atoms: # of protons = # of electrons Chemical properties of atoms # of electrons Mp ~ Mn ~ 2000 Me the mass of atom ~ the mass of nucleus Periodic table of chemical elements tabular arrangement of chemical elements based on the atomic numbers (= # of electrons = # of protons) What are we made of ? oxygen 43 kg cerium 40 mg gallium 0.7 mg carbon 16 kg barium 22 mg tellurium 0.7 mg hydrogen 7 kg iodine 20 mg yttrium 0.6 mg nitrogen 1.8 kg tin 20 mg bismuth 0.5 mg calcium 1.0 kg titanium 20 mg thallium 0.5 mg phosphorus 780 g boron 18 mg indium 0.4 mg potassium 140 g nickel 15 mg gold 0.2 mg sulphur 140 g selenium