Chemistry of Uo2 Fuel Dissolution in Relation to the Disposal of Used Nuclear Fuel
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pp-03-25-New Dots.Qxd 10/23/02 2:41 PM Page 778
pp-03-25-new dots.qxd 10/23/02 2:41 PM Page 778 778 PRAESODYMIUM PRAESODYMIUM [7440–10–0] Symbol Pr; atomic number 59; atomic weight 140.908; a lanthanide–series rare earth element; belongs to the cerium group of rare earths; electron con- figuration [Xe] 4f36s2; partially filled f subshell; valence states +3, +4; most 3+ stable oxidation state +3; electrode potential E°/V (aq) for Pr + 3e¯ ↔ Pr is –2.35 V; atomic radius 1.828 Å; first ionization potential 5.46 eV; one natu- rally–occurring isotope, Pr–141; twenty–nine artificial radioactive isotopes known in the mass range 124, 126–140 and 142–154; the longest–lived isotope Pr–143, t1/2 13.57 day, and the shortest–lived isotope Pr–124, t1/2 1.2 second. History, Occurrence, and Uses Mosander extracted from the mineral lanthana a rare earth fraction, named didymia in 1841. In 1879, Boisbaudran separated a rare earth oxide called samaria (samarium oxide) from the didymia fraction obtained from the mineral samarskite. Soon after that in 1885, Baron Auer von Welsbach iso- lated two other rare earths from didymia. He named them as praseodymia (green twin) and neodymia (new twin) after their source didymia (twin). The name praseodymium finally was assigned to this new element, derived from the two Greek words, prasios meaning green and didymos meaning twin. Praseodymium occurs in nature associated with other rare earths in a rel- atively high abundance. It is more abundant than some common metals such as silver, gold, or antimony. The average concentration of this metal in the earth’s crust is estimated to be 8.2 mg/kg. -

WO 2014/029634 Al 27 February 2014 (27.02.2014) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2014/029634 Al 27 February 2014 (27.02.2014) P O P C T (51) International Patent Classification: DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, C09C 1/02 (2006.01) HN, HR, HU, ID, IL, ΓΝ , IS, JP, KE, KG, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/EP20 13/066666 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, 8 August 2013 (08.08.2013) TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, 1218 1089.9 20 August 2012 (20.08.2012) EP UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 61/693,350 27 August 2012 (27.08.2012) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, LV, (71) Applicant: OMYA INTERNATIONAL AG [CH/CH]; MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, Baslerstrasse 42, CH-4665 Oftringen (CH). -

On the Solubility of Radium Sulfate and Carbonate
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Chalmers Publication Library THESIS FOR THE DEGREE OF LICENTIATE OF ENGINEERING On the solubility of radium sulfate and carbonate Artem Vasilyevich Matyskin Nuclear Chemistry Department of Chemistry and Chemical Engineering CHALMERS UNIVERSITY OF TECHNOLOGY Gothenburg, Sweden 2016 On the solubility of radium sulfate and carbonate © ARTEM VASILYEVICH MATYSKIN, 2016 Technical report number: 2016:04 ISSN number: 1652-943X Nuclear Chemistry Department of Chemistry and Chemical Engineering Chalmers University of Technology SE – 412 96 Göteborg Sweden Telephone +46(0)31-772 1000 Cover: Radium sulfate powder Chalmers Reproservice Gothenburg, Sweden 2016 On the solubility of radium sulfate and carbonate Artem V. Matyskin Nuclear Chemistry Department of Chemistry and Chemical Engineering Chalmers University of Technology Abstract Radium is one of the most toxic elements and its concentration in different human activities and migration from man-made wastes provokes a strong interest in environmental science. To be able to model the migration process, reliable experimental thermodynamic data of radium compounds are needed. In this work details of the safe radium source disassembly which were previously used in brachytherapy are described and different methods for conversion of RaSO4 into aqueous solution are reviewed. The method of choice included three cycles of RaSO4 heating in 1.5 M Na2CO3 up to 85 ºC, cooling and subsequent removal of supernatant. X-ray diffraction studies showed that the method allows the synthesis of amorphous RaCO3, which can be dissolved in mineral acid. Gamma spectrometric measurements showed that most of the initial RaSO4 was 210 converted into solution and that 7 ± 1 % of the initial Pb was co-precipitated with RaCO3. -

AAAS. the American Association for the Advancement of Science Was
A AAAS. The American Association for the Advancement of Science was movement through a planetary atmosphere to provide thermal protection founded in 1848 and incorporated in 1874. Its objectives are to further the to the underlying structure. See also Ablating Material. work of scientists, to facilitate cooperation among them, to foster scientific freedom and responsibility, to improve the effectiveness of science in ABRASION. All metallic and nonmetallic surfaces, no matter how promoting human welfare, to advance education in science, and to increase smooth, consist of minute serrations and ridges that induce a cutting or public understanding and appreciation for the importance and promise of tearing action when two surfaces in contact move with respect to each the methods of science in human progress. The AAAS head quarters is other. This wearing of the surfaces is termed abrasion. Undesirable abrasion in Washington, DC. Additional information on the AAAS can be found at may occur in bearings and other machine elements, but abrasion is also http://www.aaas.org/ and http://www.sciencemag.org/ . adapted to surface finishing and machining, where the material is too hard to be cut by other means, or where precision is a primary requisite. ABACA. The sclerenchyma bundles from the sheathing leaf bases of Temperature is a significant factor: friction may raise the temperature of Musa textilis, a plant closely resembling the edible banana plant. These the surface layers to the point where they become subject to chemical bundles are stripped by hand, after which they are cleaned by drawing over attack. Abrasion causes deterioration of many materials, especially of a rough knife. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

The Radiochemistry of Radium Commllleeon NUCLEAR SCIENCE
National .—. Academy d Sciences NationalI Research Council m NUCLEAR SCIENCE SERIES The Radiochemistry of Radium COMMlllEEON NUCLEAR SCIENCE B.K- Aufmn,Chdmlm R. D. Evans, Wce Chairman UnlverOity of Chfcwo ~. In.Wttute of Technology Lewis slack,Secretary NstidFtem-cllmllntil E. c.Andmrmn HertwrtC?dd&tm i.mAlumnw. laboratory Calmibla Unlvertity C.J. BmkcwM J. J. Nlckmn M Ridge MU. Iabmntm-y NEmOrial Eo9pltd (New York) RobertG.cm- IL L. Phtzma!l A& MColbWaf TcmO A~ N81icad la.tamtory ugoFam D. M Vm P+ter NaUOadwruofsmldmda Eartol RO~ Fwdmon Oeorgew.Wetbefill Urdvarslty of CUlfonda (Ian Angeles) LIAISONMRMBERS padC. Aaberwld JeromeRegean AtOmlcEwrgy-mmfmtm UfIce ofNaml Reach Ja6ePh E. Duval J. &wwd McMlflen Afr Farce OfUce of SctenUflc ~h NuUc4ml Sdence FwndaUm SIJ9COMMlllEEON RADI-RMISTRY N. E. -, Chd- J. D. Kn@t u.& NavalFwlblO@dDOfenlm ICaAlamwSelmtlfic I.atmratmy Lab0rnk3m J. M. Nielmn G. R CbqPln 06netal Electric Company (IuChiand) FlOrMn State Uutvertiw G. D. O’ftdley E. M Clark W NMge National Laboratory Rmaselaer POlytadmlc fmtitute RP. tldmman R. M“ Dimnold AtOmlc mml’w rnvistml Lawrence Radiaklm Iabm-story Pfltlllpa Petroleum COi+my (LillM FalM) A.W.Fdrf@l K P. ~rg Unlversfty of Washl@m Argmne NatfOllaf mbodmy Jerome Ndls P. c.Wevanean Brcakkav671 Natfanal fdwratory Lawrence lwnatlan LabOrafov D. N. bhiernmn Mttelb Mernorhf fmlltute coNsLaTANl J. W. Ulncbetier “~wata hlefmuti of 1%~ ,. The Radiochemistry of Radium By H. W. Kirby Mound Laboratory Monsanto Research Corporation Miamisburg, Ohio and Murrell L. Salutsky W. R. Grace & Co. Research Division Washington Research Center Clarksville, Md. kued,December19&4 Subcommitteeon Radiochemistry NationalAcademy of Sciences—NaUonal Research Council Prin&dinUSA.Price$2.25Av-dhblefromtieClear4@cmseforFederal Sciendficu@ TechnknlInfonnatlon,Nation8.1BureauofStaxhr&,U.S.De- parbnentofComme-,SPri@ield,Vir@ia. -

(12) United States Patent (10) Patent No.: US 9,725,576 B2 Sötemann (45) Date of Patent: Aug
USO09725576B2 (12) United States Patent (10) Patent No.: US 9,725,576 B2 Sötemann (45) Date of Patent: Aug. 8, 2017 (54) PROCESS FOR MANUFACTURING WHITE B03D 2203/005; B03D 2201/02: C09C PGMENT CONTAINING PRODUCTS 1/02: C09C 1/021; C04B 14/28: C04B 16/00; C08K 3/26: C08K 5/17; C08K (71) Applicant: Omya International AG. Oftringen 5/20; C08K 2003/265; C01P 2004/51: (CH) C01P 2004/61; C01P 2006/12: C01P 2006/60; C01P 2006/80 (72) Inventor: Jörg Sötemann, Villach (AT) See application file for complete search history. (73) Assignee: Omya International AG. Oftringen (CH) (56) References Cited U.S. PATENT DOCUMENTS (*) Notice: Subject to any disclaimer, the term of this patent is extended or adjusted under 35 3,990,966 A 11/1976 Stanley et al. U.S.C. 154(b) by 169 days. 4,995,965 A 2/1991 Mehaffey et al. 5,261,539 A * 11/1993 Hancock .................. BO3D 1.01 (21) Appl. No.: 14/.414,967 209,166 5,720,873 A * 2/1998 Klingberg ............ BO3D 1 0043 209,166 (22) PCT Filed: Aug. 8, 2013 8,381,915 B2 * 2/2013 Tavakkoli ................ BO3D 1.01 209,166 (86). PCT No.: PCT/EP2013/066666 8,662,311 B2 3/2014 Gane ........................ BO3D 1.01 209,166 S 371 (c)(1), 8,992,875 B2* 3/2015 Tavakkoli ............... CO1F 11/18 (2) Date: Jan. 15, 2015 423.432 2010.0040528 A1 2/2010 Tavakkoli ................ BO3D 1.01 (87) PCT Pub. No.: WO2014/029634 423 (430 2014/0048453 A1 2/2014 Birken .................... -

HYSYS OLI Interface
HYSYS® 2004.2 OLI Interface Reference Guide Copyright October 2005 Copyright © 1981-2005 by Aspen Technology, Inc. All rights reserved. Aspen Accounting.21™, Aspen ACM Model Export, Aspen ACOL™, Aspen ACX™ Upgrade to ACOL™, Aspen Adsim®, Aspen Advisor™, Aspen Aerotran®, Aspen Alarm & Event™, Aspen APLE™, Aspen Apollo™, Aspen AtOMS™, Aspen Batch and Event Extractor, Aspen Batch Plus®, Aspen Batch.21™, Aspen Batch.21™ CBT, Aspen BatchCAD™, Aspen BatchSep™, Aspen Blend Model Library™, Aspen Blend™, Aspen BP Crude Oil Database, Aspen Calc CBT, Aspen Calc™, Aspen Capable-to-Promise®, Aspen CatRef®, Aspen Chromatography®, Aspen Cim-IO Core™, Aspen Cim-IO™ for @AGlance, Aspen Cim-IO™ for ABB 1180/ 1190 via DIU, Aspen Cim-IO™ for Bailey SemAPI, Aspen Cim-IO™ for DDE, Aspen Cim-IO™ for Eurotherm Gauge via DCP, Aspen Cim-IO™ for Fisher-Rosemount Chip, Aspen Cim-IO™ for Fisher-Rosemount RNI, Aspen Cim-IO™ for Foxboro FOXAPI, Aspen Cim-IO™ for G2, Aspen Cim-IO™ for GE FANUC via HCT, Aspen Cim-IO™ for Hitachi Ex Series, Aspen Cim-IO™ for Honeywell TDC 3000 via HTL/access, Aspen Cim-IO™ for Intellution Fix, Aspen Cim-IO™ for Measurex MCN, Aspen Cim-IO™ for Measurex ODX, Aspen Cim-IO™ for Moore Apacs via Nim (RNI), Aspen Cim-IO™ for OPC, Aspen Cim-IO™ for PI, Aspen Cim- IO™ for RSLinx, Aspen Cim-IO™ for SetCim/InfoPlus-X/InfoPlus.21, Aspen Cim-IO™ for Toshiba Tosdic, Aspen Cim-IO™ for ULMA 3D, Aspen Cim-IO™ for Westinghouse, Aspen Cim-IO™ for WonderWare InTouch, Aspen Cim-IO™ for Yokogawa ACG10S, Aspen Cim-IO™ for Yokogawa EW3, Aspen Collaborative Forecasting™, -

DOCUMENT RESUME ED 124 409 Radiological Safety Handbook. 835P
DOCUMENT RESUME ED 124 409 SE 020 -603 TITLE Radiological Safety Handbook. INSTITUTION Army Ordnance Center .and School, Aberdeen Proving Ground, Md. PUB DATE [ 76 NOTE . 835p.; Occasional small print and Iighttype EDRS PRICE MF-$1.50 HC-$44.87 Plus Postage. DESCRIPTORS Armed Forces; *Instructional Materials; Lasers; measurement Inst , mar ehys-rcgr-Nuclea-r-' Warfare; *Radiatione-*Radiation Effects; *Safety; Science Education; Teaching Guides , IDENTIFIERS Army ABSTRACT Written to be used concurrently with the U.S. Army's 'Radiological Safety Course, this publication discusses the causes, sources, and detection of nuclear radiation. In. addition, the transportation and disposal of, radioactive materials are covered. The report also deals with the,safety precautions to be observed when working with lasers, microwave generators, and particle accelerators. Work problems with solutions are provided for these topics mentioned.. (CP) *********************************************************************** *- Documents acquired by ERIC include many informal unpublished * .* materials not available from other sources. ERIC:makes every effort * * to obtain the best copy. available. Nevertheless,' items of marginal* * reproducibility are often encountered and this_affects the quality * * of the microfiche and hardcopy reproductions EPIC makes available *. * via the ERIC Document Reproduction Service (EDRS). EDRS is not * * responsible for the quality of the original document. Reproductions * * supplied by EDRS are thQ best that can be made from the original. * *********************************************************************** SCOPE OF INTEREST NOTICE ft U S DEPARTMENT OF HEALTH EDUCATION & WELFARE '11,e. ERIC F.ChIy has atered NATIONAL INSTITUTE OF th.s document for rroceivh9 EDUCATION o mAS _RCPRo- In 04 14/0genlenly th,S document Ott',...E 0 ExACTLY AS REU,,E0 ;ROM 31$0 of Me, eS1 to the C'ebron- '.E SO% OR OR.;:.% ZAT014OR,Gh t,04.41eS noted to the t. -
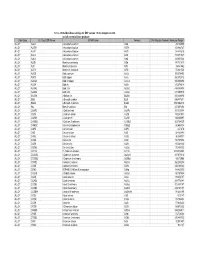
Database Full Listing
16-Nov-06 OLI Data Base Listings for ESP version 7.0.46, Analyzers 2.0.46 and all current alliance products Data Base OLI Tag (ESP) Name IUPAC Name Formula CAS Registry Number Molecular Weight ALLOY AL2U 2-Aluminum uranium Al2U 291.98999 ALLOY AL3TH 3-Aluminum thorium Al3Th 312.982727 ALLOY AL3TI 3-Aluminum titanium Al3Ti 128.824615 ALLOY AL3U 3-Aluminum uranium Al3U 318.971527 ALLOY AL4U 4-Aluminum uranium Al4U 345.953064 ALLOY ALSB Aluminum antimony AlSb 148.731537 ALLOY ALTI Aluminum titanium AlTi 74.861542 ALLOY ALTI3 Aluminum 3-titanium AlTi3 170.621536 ALLOY AUCD Gold cadmium AuCd 309.376495 ALLOY AUCU Gold copper AuCu 260.512512 ALLOY AUCU3 Gold 3-copper AuCu3 387.604492 ALLOY AUSN Gold tin AuSn 315.676514 ALLOY AUSN2 Gold 2-tin AuSn2 434.386505 ALLOY AUSN4 Gold 4-tin AuSn4 671.806519 ALLOY BA2SN 2-Barium tin Ba2Sn 393.369995 ALLOY BI2U 2-Bismuth uranium Bi2U 655.987671 ALLOY BI4U3 4-Bismuth 3-uranium Bi4U3 1550.002319 ALLOY BIU Bismuth uranium BiU 447.007294 ALLOY CA2PB 2-Calcium lead Ca2Pb 287.355988 ALLOY CA2SI 2-Calcium silicon Ca2Si 108.241501 ALLOY CA2SN 2-Calcium tin Ca2Sn 198.865997 ALLOY CA3SB2 3-Calcium 2-antimony Ca3Sb2 363.734009 ALLOY CAMG2 Calcium 2-magnesium CaMg2 88.688004 ALLOY CAPB Calcium lead CaPb 247.278 ALLOY CASI Calcium silicon CaSi 68.163498 ALLOY CASI2 Calcium 2-silicon CaSi2 96.249001 ALLOY CASN Calcium tin CaSn 158.787994 ALLOY CAZN Calcium zinc CaZn 105.468002 ALLOY CAZN2 Calcium 2-zinc CaZn2 170.858002 ALLOY CD11U 11-Cadmium uranium Cd11U 1474.536865 ALLOY CD3AS2 3-Cadmium 2-arsenic As2Cd3 487.073212 -

United States Patent (10) Patent No.: US 7,008,633 B2 Yang Et Al
USOO70O8633B2 (12) United States Patent (10) Patent No.: US 7,008,633 B2 Yang et al. (45) Date of Patent: Mar. 7, 2006 (54) LOCAL REGIONAL CHEMOTHERAPY AND 5,257,970 A 11/1993 Dougherty RADIOTHERAPY USING IN SITU 5,470,843 A 11/1995 Stahl et al. HYDROGEL 5,542.935 A 8/1996 Unger et al. 5,571,797 A 11/1996 Ohno et al. (75) Inventors: David J. Yang, Sugar Land, TX (US); 5,656.272 A 8/1997 Le et al. Dong-Fang Yu, Houston, TX (US); Ali 5,702.717. A 12/1997 Cha et al. Azhdarinia, Houston, TX (US); 5.945,100 A * 8/1999 Fick ........................ 424/93.21 Tommy L. Lee, Spring, TX (US); E. 5,977,163 A 11/1999 Li et al. Edmund Kim, Houston, TX (US) 5,989,215 A * 11/1999 Delmotte et al. ............. 604/82 s s 6,004,573 A 12/1999 Rathi et al. (73) Assignee: Board of Regents, The University of 6,117,949 A 9/2000 Rathi et al. Texas System, Austin, TX (US) FOREIGN PATENT DOCUMENTS (*) Notice: Subject to any disclaimer, the term of this JP 10236984 A2 9/1993 patent is extended or adjusted under 35 (Continued) U.S.C. 154(b) by 254 days. OTHER PUBLICATIONS (21) Appl. No.: 10/024,678 Burris III, Howard A., et al., Intratumoral cisplatin/epineph (22)22) Filled: Dec. 18,9 2001 rine-injectable gel as a palliative treatment for accessible Solid tumors: A multicenter pilot center; Head Neck Surg, (65) Prior Publication Data vol. -

Example Exercise 7.1 Classifying Compounds and Acids
Example Exercise 7.1 Classifying Compounds and Acids Classify each of the following as a binary ionic compound, ternary ionic compound, binary molecular compound, binary acid, or ternary oxyacid: (a) calcium oxide, CaO (b) sulfur dioxide, SO2 (c) silver chromate, Ag2CrO4 (d) hydrofluoric acid, HF(aq) (e) carbonic acid, H2CO3(aq) Solution We can refer to the periodic table and classify each compound or solution as follows: (a) CaO contains two elements, a metal and nonmetal. Thus, CaO is a binary ionic compound. (b) SO2 contains two elements, both nonmetals. Thus, SO2 is a binary molecular compound. (c) Ag2CrO4 contains three elements, two metals and a nonmetal. Thus, Ag2CrO4 is a ternary ionic compound. (d) HF(aq) is a compound of hydrogen and a nonmetal dissolved in water. Thus, HF(aq) is a binary acid. (e) H2CO3(aq) is a compound containing three elements, including hydrogen and oxygen, dissolved in water. Thus, H2CO3(aq) is a ternary oxyacid. Introductory Chemistry: Concepts and Critical Thinking, 6th Edition © 2011 Pearson Education, Inc. Charles H. Corwin Example Exercise 7.1 Classifying Compounds and Acids Continued Practice Exercise Classify each of the following as a binary ionic compound, ternary ionic compound, binary molecular compound, binary acid, or ternary oxyacid: (a) carbon disulfide, CS2 (b) lithium dichromate, Li2Cr2O7 (c) magnesium iodide, MgI2 (d) nitric acid, HNO3(aq) (e) hydrochloric acid, HCl(aq) Answers: (a) binary molecular compound; (b) ternary ionic compound; (c) binary ionic compound; (d) ternary oxyacid; (e) binary acid Concept Exercise How do you distinguish a binary molecular compound from a binary acid? Answer: See Appendix G.