The Radiochemistry of Radium Commllleeon NUCLEAR SCIENCE
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Amidotrizoic Acid/Barium Sulfate 1477 Imbalance Should Be Corrected Before Contrast Media Are Given
Amidotrizoic Acid/Barium Sulfate 1477 imbalance should be corrected before contrast media are given. management of adhesive small bowel obstruction;2 they allow pyloric stenosis or lesions that may predispose to obstruction. Particular care is needed in patients with multiple myeloma since identification of patients who require surgery and, although they Adequate hydration should be ensured after the procedure to pre- dehydration resulting from use of contrast media may cause pre- have not been shown to relieve obstruction, they may reduce vent severe constipation. cipitation of protein in the renal tubules, leading to anuria and length of hospital stay in patients treated without surgery. It is contra-indicated in patients with gastrointestinal perforation, fatal renal failure. 1. Murshed R, et al. Meconium ileus: a ten-year review of thirty-six and should be avoided, particularly when given rectally, in those Caution is also necessary in patients with severe hypertension, patients. Eur J Pediatr Surg 1997; 7: 275–7. at risk of perforation, such as patients with acute ulcerative colitis advanced cardiac disease, phaeochromocytoma, sickle-cell dis- 2. Abbas S, et al. Oral water soluble contrast for the management or diverticulitis and after rectal or colonic biopsy, sigmoidosco- of adhesive small bowel obstruction. Available in The Cochrane ease, or hyperthyroidism or epilepsy, and in debilitated, severely Database of Systematic Reviews; Issue 3. Chichester: John Wi- py, or radiotherapy. ill, very old, or very young patients. ley; 2007 (accessed 14/07/08). Uses and Administration Amidotrizoates and other hypertonic contrast media are neuro- Preparations Barium sulfate is used as a radiographic contrast medium toxic and should not be given intrathecally; patients with sub- (p.1474) for X-ray examination of the gastrointestinal tract in- arachnoid haemorrhage may be at risk with any intravascular BP 2008: Meglumine Amidotrizoate Injection; Sodium Amidotrizoate In- jection; volving single- or double-contrast techniques or computed tom- use. -
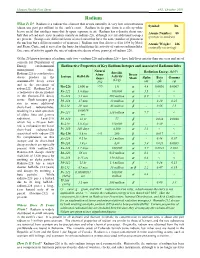
Radium What Is It? Radium Is a Radioactive Element That Occurs Naturally in Very Low Concentrations Symbol: Ra (About One Part Per Trillion) in the Earth’S Crust
Human Health Fact Sheet ANL, October 2001 Radium What Is It? Radium is a radioactive element that occurs naturally in very low concentrations Symbol: Ra (about one part per trillion) in the earth’s crust. Radium in its pure form is a silvery-white heavy metal that oxidizes immediately upon exposure to air. Radium has a density about one- Atomic Number: 88 half that of lead and exists in nature mainly as radium-226, although several additional isotopes (protons in nucleus) are present. (Isotopes are different forms of an element that have the same number of protons in the nucleus but a different number of neutrons.) Radium was first discovered in 1898 by Marie Atomic Weight: 226 and Pierre Curie, and it served as the basis for identifying the activity of various radionuclides. (naturally occurring) One curie of activity equals the rate of radioactive decay of one gram (g) of radium-226. Of the 25 known isotopes of radium, only two – radium-226 and radium-228 – have half-lives greater than one year and are of concern for Department of Energy environmental Radioactive Properties of Key Radium Isotopes and Associated Radionuclides management sites. Natural Specific Radiation Energy (MeV) Radium-226 is a radioactive Abun- Decay Isotope Half-Life Activity decay product in the dance Mode Alpha Beta Gamma (Ci/g) uranium-238 decay series (%) (α) (β) (γ) and is the precursor of Ra-226 1,600 yr >99 1.0 α 4.8 0.0036 0.0067 radon-222. Radium-228 is a radioactive decay product Rn-222 3.8 days 160,000 α 5.5 < < in the thorium-232 decay Po-218 3.1 min 290 million α 6.0 < < series. -

The Origins of the Underline As Visual Representation of the Hyperlink on the Web: a Case Study in Skeuomorphism
The Origins of the Underline as Visual Representation of the Hyperlink on the Web: A Case Study in Skeuomorphism The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Romano, John J. 2016. The Origins of the Underline as Visual Representation of the Hyperlink on the Web: A Case Study in Skeuomorphism. Master's thesis, Harvard Extension School. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:33797379 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA The Origins of the Underline as Visual Representation of the Hyperlink on the Web: A Case Study in Skeuomorphism John J Romano A Thesis in the Field of Visual Arts for the Degree of Master of Liberal Arts in Extension Studies Harvard University November 2016 Abstract This thesis investigates the process by which the underline came to be used as the default signifier of hyperlinks on the World Wide Web. Created in 1990 by Tim Berners- Lee, the web quickly became the most used hypertext system in the world, and most browsers default to indicating hyperlinks with an underline. To answer the question of why the underline was chosen over competing demarcation techniques, the thesis applies the methods of history of technology and sociology of technology. Before the invention of the web, the underline–also known as the vinculum–was used in many contexts in writing systems; collecting entities together to form a whole and ascribing additional meaning to the content. -

Vibrationally Excited Hydrogen Halides : a Bibliography On
VI NBS SPECIAL PUBLICATION 392 J U.S. DEPARTMENT OF COMMERCE / National Bureau of Standards National Bureau of Standards Bldg. Library, _ E-01 Admin. OCT 1 1981 191023 / oO Vibrationally Excited Hydrogen Halides: A Bibliography on Chemical Kinetics of Chemiexcitation and Energy Transfer Processes (1958 through 1973) QC 100 • 1X57 no. 2te c l !14 c '- — | NATIONAL BUREAU OF STANDARDS The National Bureau of Standards' was established by an act of Congress March 3, 1901. The Bureau's overall goal is to strengthen and advance the Nation's science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the Nation's physical measurement system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to promote public safety. The Bureau consists of the Institute for Basic Standards, the Institute for Materials Research, the Institute for Applied Technology, the Institute for Computer Sciences and Technology, and the Office for Information Programs. THE INSTITUTE FOR BASIC STANDARDS provides the central basis within the United States of a complete and consistent system of physical measurement; coordinates that system with measurement systems of other nations; and furnishes essential services leading to accurate and uniform physical measurements throughout the Nation's scientific community, industry, and commerce. The Institute consists of a Center for Radiation Research, an Office of Meas- urement Services and the following divisions: Applied Mathematics — Electricity — Mechanics — Heat — Optical Physics — Nuclear Sciences" — Applied Radiation 2 — Quantum Electronics 1 — Electromagnetics 3 — Time 3 1 1 and Frequency — Laboratory Astrophysics — Cryogenics . -

1-Bromopropane
Right to Know Hazardous Substance Fact Sheet Common Name: 1-BROMOPROPANE Synonyms: Propyl Bromide CAS Number: 106-94-5 Chemical Name: Propane, 1-Bromo- RTK Substance Number: 4198 Date: October 2009 Revision: March 2016 DOT Number: UN 2344 Description and Use EMERGENCY RESPONDERS >>>> SEE BACK PAGE 1-Bromopropane is a clear, colorless liquid with a sweet odor. Hazard Summary It is used in dry cleaning, and as a solvent, adhesive, and an Hazard Rating NJDHSS NFPA aerosol propellant. HEALTH 2 - FLAMMABILITY 3 - REACTIVITY 1 - Reasons for Citation 1-Bromopropane is on the Right to Know Hazardous FLAMMABLE Substance List because it is cited by the EPA, NTP, ACGIH POISONOUS GASES ARE PRODUCED IN FIRE and DOT. CONTAINERS MAY EXPLODE IN FIRE This chemical is on the Special Health Hazard Substance Hazard Rating Key: 0=minimal; 1=slight; 2=moderate; 3=serious; List. 4=severe 1-Bromopropane can affect you when inhaled and may be absorbed through the skin. 1-Bromopropane should be handled as a CARCINOGEN-- WITH EXTREME CAUTION. 1-Bromopropane may cause reproductive damage. SEE GLOSSARY ON PAGE 5. HANDLE WITH EXTREME CAUTION. Contact can irritate the skin and eyes. FIRST AID Inhaling 1-Bromopropane can irritate the nose, throat and lungs. Eye Contact Exposure can cause headache, dizziness, lightheadedness, Immediately flush with large amounts of water for at least 15 trouble concentrating, and weakness. minutes, lifting upper and lower lids. Remove contact 1-Bromopropane may damage the nervous system causing lenses, if worn, while rinsing. numbness, “pins and needles,” and/or weakness in the hands and feet. Skin Contact 1-Bromopropane may affect the liver. -

Klinkenberg Effect on Predicting and Measuring Helium Permeability in Gas Shales
International Journal of Coal Geology 123 (2014) 62–68 Contents lists available at ScienceDirect International Journal of Coal Geology journal homepage: www.elsevier.com/locate/ijcoalgeo Klinkenberg effect on predicting and measuring helium permeability in gas shales Mahnaz Firouzi, Khalid Alnoaimi, Anthony Kovscek, Jennifer Wilcox ⁎ Department of Energy Resources Engineering, Stanford University, Stanford, CA 94305-2220, USA article info abstract Article history: To predict accurately gas-transport in shale systems, it is important to study the transport phenomena of a non- Received 21 June 2013 adsorptive gas such as helium to investigate gas “slippage”. Non-equilibrium molecular dynamics (NEMD) sim- Received in revised form 24 September 2013 ulations have been carried out with an external driving force imposed on the 3-D carbon pore network generated Accepted 24 September 2013 atomistically using the Voronoi tessellation method, representative of the carbon-based kerogen porous struc- Available online 1 October 2013 ture of shale, to investigate helium transport and predict Klinkenberg parameters. Simulations are conducted to determine the effect of pressure on gas permeability in the pore network structure. In addition, pressure Keywords: Klinkenberg pulse decay experiments have been conducted to measure the helium permeability and Klinkenberg parameters Molecular modeling of a shale core plug to establish a comparison between permeability measurements in the laboratory and the per- Measurement meability predictions using NEMD simulations. The results indicate that the gas permeability, obtained from Helium pulse-decay experiments, is approximately two orders of magnitude greater than that calculated by the MD sim- Permeability ulation. The experimental permeability, however, is extracted from early-time pressure profiles that correspond Shale to convective flow in shale macropores (N50 nm). -

The National Drugs List
^ ^ ^ ^ ^[ ^ The National Drugs List Of Syrian Arab Republic Sexth Edition 2006 ! " # "$ % &'() " # * +$, -. / & 0 /+12 3 4" 5 "$ . "$ 67"5,) 0 " /! !2 4? @ % 88 9 3: " # "$ ;+<=2 – G# H H2 I) – 6( – 65 : A B C "5 : , D )* . J!* HK"3 H"$ T ) 4 B K<) +$ LMA N O 3 4P<B &Q / RS ) H< C4VH /430 / 1988 V W* < C A GQ ") 4V / 1000 / C4VH /820 / 2001 V XX K<# C ,V /500 / 1992 V "!X V /946 / 2004 V Z < C V /914 / 2003 V ) < ] +$, [2 / ,) @# @ S%Q2 J"= [ &<\ @ +$ LMA 1 O \ . S X '( ^ & M_ `AB @ &' 3 4" + @ V= 4 )\ " : N " # "$ 6 ) G" 3Q + a C G /<"B d3: C K7 e , fM 4 Q b"$ " < $\ c"7: 5) G . HHH3Q J # Hg ' V"h 6< G* H5 !" # $%" & $' ,* ( )* + 2 ا اوا ادو +% 5 j 2 i1 6 B J' 6<X " 6"[ i2 "$ "< * i3 10 6 i4 11 6! ^ i5 13 6<X "!# * i6 15 7 G!, 6 - k 24"$d dl ?K V *4V h 63[46 ' i8 19 Adl 20 "( 2 i9 20 G Q) 6 i10 20 a 6 m[, 6 i11 21 ?K V $n i12 21 "% * i13 23 b+ 6 i14 23 oe C * i15 24 !, 2 6\ i16 25 C V pq * i17 26 ( S 6) 1, ++ &"r i19 3 +% 27 G 6 ""% i19 28 ^ Ks 2 i20 31 % Ks 2 i21 32 s * i22 35 " " * i23 37 "$ * i24 38 6" i25 39 V t h Gu* v!* 2 i26 39 ( 2 i27 40 B w< Ks 2 i28 40 d C &"r i29 42 "' 6 i30 42 " * i31 42 ":< * i32 5 ./ 0" -33 4 : ANAESTHETICS $ 1 2 -1 :GENERAL ANAESTHETICS AND OXYGEN 4 $1 2 2- ATRACURIUM BESYLATE DROPERIDOL ETHER FENTANYL HALOTHANE ISOFLURANE KETAMINE HCL NITROUS OXIDE OXYGEN PROPOFOL REMIFENTANIL SEVOFLURANE SUFENTANIL THIOPENTAL :LOCAL ANAESTHETICS !67$1 2 -5 AMYLEINE HCL=AMYLOCAINE ARTICAINE BENZOCAINE BUPIVACAINE CINCHOCAINE LIDOCAINE MEPIVACAINE OXETHAZAINE PRAMOXINE PRILOCAINE PREOPERATIVE MEDICATION & SEDATION FOR 9*: ;< " 2 -8 : : SHORT -TERM PROCEDURES ATROPINE DIAZEPAM INJ. -

22Sra 226Ra and 222Rn in ' SOUTH CAROLINA GROUND WATER
Report No. 95 WRRI 22sRa 226Ra AND 222Rn IN ' SOUTH CAROLINA GROUND WATER: MEASUR~MEN-T TECHNIQUES AND ISOTOPE RELATIONSHIPS GlANNll\li FCUN~On o;: t,GR!CUL.TU~~tcoNOMlC2' ~~J.i\2Y J\li·iU ('lJ ~C 4· 1982 WATER RESOURCES RESEARCH INSTITUTE CLEMSON UNIVERSITY Clemson, .South Carolina JANUARY 1982 Water Resources Research Institute Clemson University Clemson, South Carolina 29631 ! 228Ra, 226Ra AND 222 Rn IN SOUTH CAROLINA GROUND WATER: MEASUREMENT TECHNIQUES AND ISOTOPE RELATIONSHIPS by Jacqueline Michel, Philip T. King and Willard S. Moore Department of Geology University of South Carolina Columbia, South Carolina 29208 The work upon which this puhlication is based was supported in part by funds provided by the Office of Water Research and Technology, project !!' No. OWRT-B-127-SC, U.S. Department of the Interior, fvashington, D. C., as authorized by the Water Research and Development Act of 1978 (PL95-467). r Project agreement No. 14-34-0001-9158 Period of Investigation: October 1979 - September 1980 Clemson University Water Resources Research Institute Technical Report No. 95 Contents of this publication do not necessarily reflect the views and policies of the Office of Water Research and Technology, U.S. Department of Interior, nor does mention of trade names or cormnercial products con stitute their endorsement or recommendation for use by the U.S. Goverrtment. ACKNOWLEDGEMENTS Much appreciation goes to Lewis Shaw, Rossie Stephens, Jim Ferguson and a large number of district personnel at the South Carolina Department of Health and Environmental Control who assisted us in field collection and provided much helpful information. Dr. -

Cool Reaction the Endothermic Reaction Between SCIENTIFIC Barium Hydroxide and Ammonium Thiocyanate
Cool Reaction The Endothermic Reaction Between SCIENTIFIC Barium Hydroxide and Ammonium Thiocyanate Introduction Many reactions produce heat, in fact when people think of chemical reactions, heat production is often expected. However, endothermic reactions, reactions which consume heat, can be just as exciting. One of the most striking examples of this is when the solids barium hydroxide and ammonium thiocyanate are mixed together in a beaker. Materials Ammonium thiocyanate, NH4SCN, 10 g Stirring rod Barium hydroxide octahydrate, Ba(OH)28H2O, 20 g Thermometer graduated to at least –30 °C Erlenmeyer flask, small, with stopper, or a 50-mL beaker Safety Precautions Barium salts are toxic by ingestion. Ammonium thiocyanate is also toxic by ingestion. Use caution when handling the beaker or flask. Use tongs if available. The temperatures involved are cold enough to freeze skin. Ammonia vapor is very irritating to eyes and the respiratory tract. Do not allow students to inhale this gas. Wear chemical splash goggles, chemical-resistant gloves, and a chemical-resistant apron. Please review current Material Safety Data Sheets for additional safety, handling, and disposal information. Procedure 1. Transfer 20 g of barium hydroxide and 10 g of ammonium thiocyanate to a flask and mix with a glass or plastic stirring rod. 2. In less than two minutes the solids become liquid. A thermometer placed in the mixture shows the temperature falling far below freezing. An ammonia odor is evident to all who are near the flask. 3. Place the flask in a small puddle of water and your students will clearly see just how cool this reaction is; the water will freeze the flask to the counter top. -

Writing Total and Net Ionic Equations
WRITING TOTAL AND NET IONIC EQUATIONS http://www.csun.edu/~hcchm001/FreshChemHandouts.html 1. Write the overall equation including the correct designations for the physical state of the substances (s, l, g, aq). Balance this equation. Most of these kinds of equations are double displacement reactions: AX + BY 6 AY + BX 2. For the total ionic equations, write strong electrolytes in solution in the form of aqueous ions. (a) Strong acids. The common strong acids and their aqueous ions are: HI Hydroiodic acid H+-(aq) + I (aq) HBr Hydrobromic acid H+-(aq) + Br (aq) HCl Hydrochloric acid H+-(aq) + Cl (aq) +- HNO33Nitric acid H (aq) + NO (aq) +- HClO44Perchloric acid H (aq) + ClO (aq) +-2 H24SO Sulfuric acid 2 H (aq) + SO4(aq) (b) Strong bases. Strong bases are the hydroxides of the alkali (Group IA) and alkaline earth (Group IIA) metals ions which are sufficiently soluble. The common strong bases and their aqueous ions are: LiOH Lithium hydroxide Li+-(aq) + OH (aq) NaOH Sodium hydroxide Na+-(aq) + OH (aq) KOH Potassium hydroxide K+-(aq) + OH (aq) +2 - Sr(OH)2Strontium hydroxide Sr (aq) + 2 OH (aq) +2 - Ba(OH)2 Barium hydroxide Ba (aq) + 2 OH (aq) (c) Soluble salts. Determinations of the solubility of a salt may be made by reference to SOLUBILITIES OF IONIC COMPOUNDS. Soluble salts are written as their aqueous ions: NaCl(aq) Sodium chloride Na+-(aq) + Cl (aq) +-2 K24SO (aq) Potassium sulfate 2 K (aq) + SO4(aq) +-2 Li23CO (aq) Lithium carbonate 2 Li (aq) + CO3(aq) +-3 Na34PO (aq) Sodium phosphate 3 Na (aq) + PO4(aq) +-2 (NH42) SO4(aq) Ammonium sulfate 2 NH4(aq) + SO4 (aq) 3. -

Carrier Dynamics in Single Luminescent Silicon Quantum Dots
Carrier Dynamics in Single Luminescent Silicon Quantum dots Fatemeh Sangghaleh PhD Thesis Information and Communication Technology KTH Royal Institute of Technology Stockholm, Sweden 2015 TRITA-ICT 2015:09 KTH School of Information and Communication Technology SE-164 40, ISBN 978-91-7595-665-7 Kista, SWEDEN A dissertation submitted to KTH Royal Institute of Technology, Stockholm, Sweden, in partial fulfillment of requirements for the degree of Teknologie Doktor (Doctor of Philosophy). The defense will take place on October 23 2015 at 10.00 a. m. at Sal C, KTH- Electrum, Kistagången 16, Kista. © Fatemeh Sangghaleh, October 2015. Printed by: Universitetsservice US-AB Abstract Bulk silicon as an indirect bandgap semiconductor is a poor light emitter. In contrast, silicon nanocrystals (Si NCs) exhibit strong emission even at room temperature, discovered initially at 1990 for porous silicon by Leigh Canham. This can be explained by the indirect to quasi-direct bandgap modification of nano-sized silicon according to the already well-established model of quantum confinement. In the absence of deep understanding of numerous fundamental optical properties of Si NCs, it is essential to study their photoluminescence (PL) characteristics at the single-dot level. This thesis presents new experimental results on various photoluminescence mechanisms in single silicon quantum dots (Si QDs). The visible and near infrared emission of Si NCs are believed to originate from the band-to-band recombination of quantum confined excitons. However, the mechanism of such process is not well understood yet. Through time-resolved PL decay spectroscopy of well-separated single Si QDs, we first quantitatively established that the PL decay character varies from dot-to-dot and the individual lifetime dispersion results in the stretched exponential decays of ensembles. -

The Radiochemistry of Tungsten
National Academy of Sciences !National Research Council NUCLEAR SCIENCE SERIES The Radiochemistry of Tungsten — ...—- L. F. C URTISS,Chairman ROBLEY D. EVANS, Vice Chairman NationalBureau ofStandards MassachusettsInstituteofTechnology J.A. DeJUREN, Secretary WestinghouseElectricCorporation C. J.BORKOWSKI J.W. IRVINE,JR. Oak RidgeNationalLaboratory MassachusettsI&tituteofTechnology ROBERT G. COCHRAN E. D. KLEMA Texas Agriculturaland Mechanical NorthwesternUniversity College W. WAYNE MEINKE SAMUEL EPSTEIN UniversityofMichigan CaliforniaInstituteofTechnology J.J.NICKSON Memorial Hospital,New York U. FANO NationalBureau ofStandards ROBERT L. PLATZMAN Laboratoirede Chimie Physique HERBERT GOLDSTEIN NuclearDevelopmentCorporationof D. M. VAN PATTER America BartolResearch Foundation LIAISON MEMBERS PAUL C. AEBERSOLD CHARLES K. REED Atomic Energy Commission U. S.Air Force J.HOWARD McMILLEN WILLIAM E. WRIGHT NationalScienceFoundation OfficeofNavalResearch SUBCOMMITTEE ON RADIOCHEMISTRY W. WAYNE MEINKE, Chai~man HAROLD KIRBY UniversityofMichigan Mound Laboratory GREGORY R. CHOPPIN GEORGE LEDDICOTTE FloridaStateUniversity Oak RidgeNationalLaboratory GEORGE A. COWAN JULIAN NIELSEN Los Alamos ScientificLaboratory HanfordLaboratories ARTHUR W. FAIRHALL ELLIS P. STEINBERG UniversityofWashington Argonne NationalLaboratory JEROME HUDIS PETER C. STEVENSON BrookhavenNationalLaboratory UniversityofCalifornia(Livermore) EARL HYDE LEO YAFFE UniversityofC slifornia(Berkeley) McGillUniversity CONSULTANTS NATHAN BALLOU JAMES DeVOE NavalRadiologicalDefenseLaboratory