Experimental Γ Ray Spectroscopy and Investigations of Environmental Radioactivity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
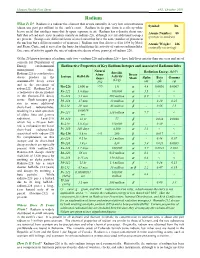
Radium What Is It? Radium Is a Radioactive Element That Occurs Naturally in Very Low Concentrations Symbol: Ra (About One Part Per Trillion) in the Earth’S Crust
Human Health Fact Sheet ANL, October 2001 Radium What Is It? Radium is a radioactive element that occurs naturally in very low concentrations Symbol: Ra (about one part per trillion) in the earth’s crust. Radium in its pure form is a silvery-white heavy metal that oxidizes immediately upon exposure to air. Radium has a density about one- Atomic Number: 88 half that of lead and exists in nature mainly as radium-226, although several additional isotopes (protons in nucleus) are present. (Isotopes are different forms of an element that have the same number of protons in the nucleus but a different number of neutrons.) Radium was first discovered in 1898 by Marie Atomic Weight: 226 and Pierre Curie, and it served as the basis for identifying the activity of various radionuclides. (naturally occurring) One curie of activity equals the rate of radioactive decay of one gram (g) of radium-226. Of the 25 known isotopes of radium, only two – radium-226 and radium-228 – have half-lives greater than one year and are of concern for Department of Energy environmental Radioactive Properties of Key Radium Isotopes and Associated Radionuclides management sites. Natural Specific Radiation Energy (MeV) Radium-226 is a radioactive Abun- Decay Isotope Half-Life Activity decay product in the dance Mode Alpha Beta Gamma (Ci/g) uranium-238 decay series (%) (α) (β) (γ) and is the precursor of Ra-226 1,600 yr >99 1.0 α 4.8 0.0036 0.0067 radon-222. Radium-228 is a radioactive decay product Rn-222 3.8 days 160,000 α 5.5 < < in the thorium-232 decay Po-218 3.1 min 290 million α 6.0 < < series. -

Bellucci Et Al. 2016 Trinitite Revised V Final
1 Direct Pb isotopic analysis of a nuclear fallout debris particle from the 2 Trinity nuclear test 3 4 5 Jeremy J. Bellucci1*, Joshua F. Snape1, Martin W. Whitehouse1, Alexander A. Nemchin1,2 6 7 *Corresponding author, email address: [email protected] 8 9 1Department of Geosciences, Swedish Museum of Natural History, SE-104 05 10 Stockholm, Sweden 11 2 Department of Applied Geology, Curtin University, Perth, WA 6845, Australia 12 13 Abstract 14 15 The Pb isotope composition of a nuclear fallout debris particle has been directly 16 measured in post-detonation materials produced during the Trinity nuclear test by a 17 secondary ion mass spectrometry (SIMS) scanning ion image technique (SII). This 18 technique permits the visual assessment of the spatial distribution of Pb and can be used 19 to obtain full Pb isotope compositions in user-defined regions in a 70 µm x 70 µm 20 analytical window. In conjunction with BSE and EDS mapping of the same particle, the 21 Pb measured in this fallout particle cannot be from a major phase in the precursor arkosic 22 sand. Similarly, the Pb isotope composition of the particle is resolvable from the 23 surrounding glass at the 2σ uncertainty level. The Pb isotope composition measured in 24 the particle here is in excellent agreement with that inferred from measurements of green 25 and red trinitite, suggesting that these types of particles are responsible for the Pb isotope 26 compositions measured in both trinitite glasses. 27 28 29 Keywords: 30 31 Trinitite, Trinity Test, Post Detonation Nuclear Forensics, Pb isotopes, SIMS, Scanning 32 Ion Imaging, Fallout Debris, Forensics 33 34 35 Introduction 36 37 In the event of a non-state sanctioned nuclear attack, forensic investigations will 38 be necessary to determine the provenance of the weapon and the materials used in 39 construction of the device. -

22Sra 226Ra and 222Rn in ' SOUTH CAROLINA GROUND WATER
Report No. 95 WRRI 22sRa 226Ra AND 222Rn IN ' SOUTH CAROLINA GROUND WATER: MEASUR~MEN-T TECHNIQUES AND ISOTOPE RELATIONSHIPS GlANNll\li FCUN~On o;: t,GR!CUL.TU~~tcoNOMlC2' ~~J.i\2Y J\li·iU ('lJ ~C 4· 1982 WATER RESOURCES RESEARCH INSTITUTE CLEMSON UNIVERSITY Clemson, .South Carolina JANUARY 1982 Water Resources Research Institute Clemson University Clemson, South Carolina 29631 ! 228Ra, 226Ra AND 222 Rn IN SOUTH CAROLINA GROUND WATER: MEASUREMENT TECHNIQUES AND ISOTOPE RELATIONSHIPS by Jacqueline Michel, Philip T. King and Willard S. Moore Department of Geology University of South Carolina Columbia, South Carolina 29208 The work upon which this puhlication is based was supported in part by funds provided by the Office of Water Research and Technology, project !!' No. OWRT-B-127-SC, U.S. Department of the Interior, fvashington, D. C., as authorized by the Water Research and Development Act of 1978 (PL95-467). r Project agreement No. 14-34-0001-9158 Period of Investigation: October 1979 - September 1980 Clemson University Water Resources Research Institute Technical Report No. 95 Contents of this publication do not necessarily reflect the views and policies of the Office of Water Research and Technology, U.S. Department of Interior, nor does mention of trade names or cormnercial products con stitute their endorsement or recommendation for use by the U.S. Goverrtment. ACKNOWLEDGEMENTS Much appreciation goes to Lewis Shaw, Rossie Stephens, Jim Ferguson and a large number of district personnel at the South Carolina Department of Health and Environmental Control who assisted us in field collection and provided much helpful information. Dr. -

Chapter 1.2: Transformation Kinetics
Chapter 1: Radioactivity Chapter 1.2: Transformation Kinetics 200 NPRE 441, Principles of Radiation Protection Chapter 1: Radioactivity http://www.world‐nuclear.org/info/inf30.html 201 NPRE 441, Principles of Radiation Protection Chapter 1: Radioactivity Serial Transformation In many situations, the parent nuclides produce one or more radioactive offsprings in a chain. In such cases, it is important to consider the radioactivity from both the parent and the daughter nuclides as a function of time. • Due to their short half lives, 90Kr and 90Rb will be completely transformed, results in a rapid building up of 90Sr. • 90Y has a much shorter half‐life compared to 90Sr. After a certain period of time, the instantaneous amount of 90Sr transformed per unit time will be equal to that of 90Y. •Inthiscase,90Y is said to be in a secular equilibrium. 202 NPRE 441, Principles of Radiation Protection Chapter 1: Radioactivity Exponential DecayTransformation Kinetics • Different isotopes are characterized by their different rate of transformation (decay). • The activity of a pure radionuclide decreases exponentially with time. For a given sample, the number of decays within a unit time window around a given time t is a Poisson random variable, whose expectation is given by t Q Q0e •Thedecayconstant is the probability of a nucleus of the isotope undergoing a decay within a unit period of time. 203 NPRE 441, Principles of Radiation Protection Chapter 1: Radioactivity Why Exponential Decay? The decay rate, A, is given by Separate variables in above equation, we have The decay constant is the probability of a nucleus of the isotope undergoing a decay within a unit period of time. -

Understanding Variation in Partition Coefficient, Kd, Values: Volume II
United States Office of Air and Radiation EPA 402-R-99-004B Environmental Protection August 1999 Agency UNDERSTANDING VARIATION IN PARTITION COEFFICIENT, Kd, VALUES Volume II: Review of Geochemistry and Available Kd Values for Cadmium, Cesium, Chromium, Lead, Plutonium, Radon, Strontium, Thorium, Tritium (3H), and Uranium UNDERSTANDING VARIATION IN PARTITION COEFFICIENT, Kd, VALUES Volume II: Review of Geochemistry and Available Kd Values for Cadmium, Cesium, Chromium, Lead, Plutonium, Radon, Strontium, Thorium, Tritium (3H), and Uranium August 1999 A Cooperative Effort By: Office of Radiation and Indoor Air Office of Solid Waste and Emergency Response U.S. Environmental Protection Agency Washington, DC 20460 Office of Environmental Restoration U.S. Department of Energy Washington, DC 20585 NOTICE The following two-volume report is intended solely as guidance to EPA and other environmental professionals. This document does not constitute rulemaking by the Agency, and cannot be relied on to create a substantive or procedural right enforceable by any party in litigation with the United States. EPA may take action that is at variance with the information, policies, and procedures in this document and may change them at any time without public notice. Reference herein to any specific commercial products, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government. ii FOREWORD Understanding the long-term behavior of contaminants in the subsurface is becoming increasingly more important as the nation addresses groundwater contamination. Groundwater contamination is a national concern as about 50 percent of the United States population receives its drinking water from groundwater. -

R-Process Elements from Magnetorotational Hypernovae
r-Process elements from magnetorotational hypernovae D. Yong1,2*, C. Kobayashi3,2, G. S. Da Costa1,2, M. S. Bessell1, A. Chiti4, A. Frebel4, K. Lind5, A. D. Mackey1,2, T. Nordlander1,2, M. Asplund6, A. R. Casey7,2, A. F. Marino8, S. J. Murphy9,1 & B. P. Schmidt1 1Research School of Astronomy & Astrophysics, Australian National University, Canberra, ACT 2611, Australia 2ARC Centre of Excellence for All Sky Astrophysics in 3 Dimensions (ASTRO 3D), Australia 3Centre for Astrophysics Research, Department of Physics, Astronomy and Mathematics, University of Hertfordshire, Hatfield, AL10 9AB, UK 4Department of Physics and Kavli Institute for Astrophysics and Space Research, Massachusetts Institute of Technology, Cambridge, MA 02139, USA 5Department of Astronomy, Stockholm University, AlbaNova University Center, 106 91 Stockholm, Sweden 6Max Planck Institute for Astrophysics, Karl-Schwarzschild-Str. 1, D-85741 Garching, Germany 7School of Physics and Astronomy, Monash University, VIC 3800, Australia 8Istituto NaZionale di Astrofisica - Osservatorio Astronomico di Arcetri, Largo Enrico Fermi, 5, 50125, Firenze, Italy 9School of Science, The University of New South Wales, Canberra, ACT 2600, Australia Neutron-star mergers were recently confirmed as sites of rapid-neutron-capture (r-process) nucleosynthesis1–3. However, in Galactic chemical evolution models, neutron-star mergers alone cannot reproduce the observed element abundance patterns of extremely metal-poor stars, which indicates the existence of other sites of r-process nucleosynthesis4–6. These sites may be investigated by studying the element abundance patterns of chemically primitive stars in the halo of the Milky Way, because these objects retain the nucleosynthetic signatures of the earliest generation of stars7–13. -

Journal of Radioanalytical and Nuclear Chemistry
J Radioanal Nucl Chem (2013) 298:993–1003 DOI 10.1007/s10967-013-2497-8 A multi-method approach for determination of radionuclide distribution in trinitite Christine Wallace • Jeremy J. Bellucci • Antonio Simonetti • Tim Hainley • Elizabeth C. Koeman • Peter C. Burns Received: 21 January 2013 / Published online: 13 April 2013 Ó Akade´miai Kiado´, Budapest, Hungary 2013 Abstract The spatial distribution of radiation within trin- The results from this study indicate that the device-related ititethinsectionshavebeenmappedusingalphatrack radionuclides were preferentially incorporated into the radiography and beta autoradiography in combination with glassy matrix in trinitite. optical microscopy and scanning electron microscopy. Alpha and beta maps have identified areas of higher activity, Keywords Trinitite Á Nuclear forensics Á Radionuclides Á and these are concentrated predominantly within the surfi- Fission and activation products Á Laser ablation inductively cial glassy component of trinitite. Laser ablation-inductively coupled plasma mass spectrometry coupled plasma mass spectrometry (LA-ICP-MS) analyses conducted at high spatial resolution yield weighted average 235U/238Uand240Pu/239Pu ratios of 0.00718 ± 0.00018 (2r) Introduction and 0.0208 ± 0.0012 (2r), respectively, and also reveal the presence of some fission (137Cs) and activation products Nuclear proliferation and terrorism are arguably the gravest (152,154Eu). The LA-ICP-MS results indicate positive cor- of threats to the security of any nation. The ability to relations between Pu ion signal intensities and abundances decipher forensic signatures in post-detonation nuclear of Fe, Ca, U and 137Cs. These trends suggest that Pu in debris is essential, both to provide a deterrence and to trinitite is associated with remnants of certain chemical permit a response to an incident. -

Lightning Strikes As a Major Facilitator of Prebiotic Phosphorus Reduction on Early Earth ✉ Benjamin L
ARTICLE https://doi.org/10.1038/s41467-021-21849-2 OPEN Lightning strikes as a major facilitator of prebiotic phosphorus reduction on early Earth ✉ Benjamin L. Hess 1,2,3 , Sandra Piazolo 2 & Jason Harvey2 When hydrated, phosphides such as the mineral schreibersite, (Fe,Ni)3P, allow for the synthesis of important phosphorus-bearing organic compounds. Such phosphides are com- mon accessory minerals in meteorites; consequently, meteorites are proposed to be a main 1234567890():,; source of prebiotic reactive phosphorus on early Earth. Here, we propose an alternative source for widespread phosphorus reduction, arguing that lightning strikes on early Earth potentially formed 10–1000 kg of phosphide and 100–10,000 kg of phosphite and hypo- phosphite annually. Therefore, lightning could have been a significant source of prebiotic, reactive phosphorus which would have been concentrated on landmasses in tropical regions. Lightning strikes could likewise provide a continual source of prebiotic reactive phosphorus independent of meteorite flux on other Earth-like planets, potentially facilitating the emer- gence of terrestrial life indefinitely. 1 Department of Earth and Planetary Sciences, Yale University, New Haven, CT, USA. 2 School of Earth and Environment, Institute of Geophysics and Tectonics, The University of Leeds, Leeds, UK. 3 Department of Geology and Environmental Science, Wheaton College, Wheaton, IL, USA. ✉ email: [email protected] NATURE COMMUNICATIONS | (2021) 12:1535 | https://doi.org/10.1038/s41467-021-21849-2 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-021-21849-2 ife on Earth likely originated by 3.5 Ga1 with carbon isotopic Levidence suggesting as early as 3.8–4.1 Ga2,3. -

Nuclide Safety Data Sheet Iron-59
59 Nuclide Safety Data Sheet 59 Iron-59 Fe www.nchps.org Fe I. PHYSICAL DATA 1 Gammas/X-rays: 192 keV (3% abundance); 1099 keV (56%), 1292 keV (44%) Radiation: 1 Betas: 131 keV (1%), 273 keV (46%), 466 keV (53%) 2 Gamma Constant: 0.703 mR/hr per mCi @ 1.0 meter [1.789E-4 mSv/hr per MBq @ 1.0 meter] 1 Half-Life [T½] : Physical T½: 44.5 day 3 Biological T½: ~1.7 days (ingestion) ; much longer for small fraction Effective T½: ~1.7 days (varies; also small fraction retained much longer) Specific Activity: 4.97E5 Ci/g [1.84E15 Bq/g] max.1 II. RADIOLOGICAL DATA Radiotoxicity4: 1.8E-9 Sv/Bq (67 mrem/uCi) of 59Fe ingested [CEDE] 4.0E-9 Sv/Bq (150 mrem/uCi) of 59Fe inhaled [CEDE] Critical Organ: Spleen1, [blood] Intake Routes: Ingestion, inhalation, puncture, wound, skin contamination (absorption); Radiological Hazard: Internal Exposure; Contamination III. SHIELDING Half Value Layer [HVL] Tenth Value Layer [TVL] Lead [Pb] 15 mm (0.59 inches) 45 mm (1.8 inches) Steel 35 mm (1.4 inches) 91 mm (3.6 inches) The accessible dose rate should be background but must be < 2 mR/hr IV. DOSIMETRY MONITORING - Always wear radiation dosimetry monitoring badges [body & ring] whenever handling 59Fe V. DETECTION & MEASUREMENT Portable Survey Meters: Geiger-Mueller [e.g. PGM] to assess shielding effectiveness & contamination Wipe Test: Liquid Scintillation Counter, Gamma Counter VI. SPECIAL PRECAUTIONS • Avoid skin contamination [absorption], ingestion, inhalation, & injection [all routes of intake] • Use shielding (lead) to minimize exposure while handling of 59Fe • Use tools (e.g. -

3 Gamma-Ray Detectors
3 Gamma-Ray Detectors Hastings A Smith,Jr., and Marcia Lucas S.1 INTRODUCTION In order for a gamma ray to be detected, it must interact with matteu that interaction must be recorded. Fortunately, the electromagnetic nature of gamma-ray photons allows them to interact strongly with the charged electrons in the atoms of all matter. The key process by which a gamma ray is detected is ionization, where it gives up part or all of its energy to an electron. The ionized electrons collide with other atoms and liberate many more electrons. The liberated charge is collected, either directly (as with a proportional counter or a solid-state semiconductor detector) or indirectly (as with a scintillation detector), in order to register the presence of the gamma ray and measure its energy. The final result is an electrical pulse whose voltage is proportional to the energy deposited in the detecting medhtm. In this chapter, we will present some general information on types of’ gamma-ray detectors that are used in nondestructive assay (NDA) of nuclear materials. The elec- tronic instrumentation associated with gamma-ray detection is discussed in Chapter 4. More in-depth treatments of the design and operation of gamma-ray detectors can be found in Refs. 1 and 2. 3.2 TYPES OF DETECTORS Many different detectors have been used to register the gamma ray and its eneqgy. In NDA, it is usually necessary to measure not only the amount of radiation emanating from a sample but also its energy spectrum. Thus, the detectors of most use in NDA applications are those whose signal outputs are proportional to the energy deposited by the gamma ray in the sensitive volume of the detector. -

Analysis of Isotopes of Radium, Uranium and Thorium. 210Pb-210Po
Analysis of isotopes of radium, uranium and thorium. 210Pb-210Po Per Roos Risø National Laboratory for Sustainable Energy, Technical university of Denmark Radium isotopes Adsorption of Ra on MnO2 & direct alpha spectrometry Coating of polyamid (Nylon) with MnO2 (KMnO4 solution) ……half-life of Ra-224 is 88h Very thin MnO2 layer, poor yield but excellent resolution Electrodeposited sources Direct 20 days 6 months Analysis of 226Ra in water • Weigh up a suitable amount of water (10 litre is mostly sufficient). • Add HNO3 to pH 1-2. • Add 133Ba tracer (ca 10 Bq). • Add about 13 ml of a 0.2M KMnO4. • Adjust pH to about 8-9 with ammonia. • Add 15 ml 0.3M MnCl2 and stir for some 5 minutes. • Adjust with KMnO4 or MnCl2 to make the sample water phase slightly pink. • Collect the precipitate and centrifuge • Dissolve precipitate in 1M HCl-1%H2O2. • Add about 2g K2SO4+1ml H2SO4 and dilute to 100 ml with water. • Heat the sample to dissolve the K2SO4 completely. • Add 30 mg Pb2+. Heat for 20 minutes and stir. • Centrifuge the precipitate and discard supernate. • Wash precipitate with 100 ml of a solution of 0.2M H2SO4 and 0.1 M K2SO4 • Centrifuge the precipitate and discard supernate • Dissolve the PbSO4-precipitate with 3ml 0.1M EDTA at pH 10. • Transfer the solution to the scintillation vial (low diffusion PE-vial). • Wash centrifuge tube with 2 ml 0.1M EDTA and transfer to Scintillation vial. • Add 10ml OptiFluor 0 and fill with deionised water to total 20 ml. • Allow for ingrowth of 222Rn during 3 weeks and measure by LSQ. -

Sources of Gamma Radiation in a Reactor Core Matts Roas
AE-19 Sources of gamma radiation in a reactor core Matts Roas AKTIEBOLAGET ATOMENERGI STOCKHOLM • S\\ HDJtN • 1959 AE-19 ERRATUM The spectrum in Fig. 3 has erroneously been normalized to 7. 4 MeV/capture. The correct spectrum can be found by mul- tiplying the ordinate by 0. 64. AE-19 Sources of gamma radiation in a reactor core Matts Roos Summary: - In a thermal reactor the gamma ray sources of importance for shielding calculations and related aspects are 1) fission, 2) decay of fission products, 3) capture processes in fuel, poison and other materials, 4) inelastic scattering in the fuel and 5) decay of capture products. The energy release and the gamma ray spectra of these sources have been compiled or estimated from the latest information available, and the results are presented in a general way to permit 235 application to any thermal reactor, fueled with a mixture of U and 238 U • As an example the total spectrum and the spectrum of radiation escaping from a fuel rod in the Swedish R3-reactor are presented. Completion of manuscript April 1959 Printed Maj 1959 LIST OF CONTENTS Page Introduction ........... 1 1. Prompt fis sion gamma rays i 2. Fission product gamma rays 2 3. Uranium capture gamma rays 4 O -2 Q 4. U inelastic scattering gamma rays 5 5. Gamma rays from capture in poison, construction materials and moderator .....*•»..•........ 8 6. Gamma rays from disintegration of capture products. 8 7. Total gamma spectra. Application to the Swedish R3 -reactor 9 SOURCES OF GAMMA RADIATION IN A REACTOR CORE.