Molecular Profiling of Soil Animal Diversity in Natural Ecosystems
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![Species Variability and Connectivity in the Deep Sea: Evaluating Effects of Spatial Heterogeneity and Hydrodynamic Effects]](https://docslib.b-cdn.net/cover/5381/species-variability-and-connectivity-in-the-deep-sea-evaluating-effects-of-spatial-heterogeneity-and-hydrodynamic-effects-615381.webp)
Species Variability and Connectivity in the Deep Sea: Evaluating Effects of Spatial Heterogeneity and Hydrodynamic Effects]
Supplementary material for [L Lins], [2016], [Species variability and connectivity in the deep sea: evaluating effects of spatial heterogeneity and hydrodynamic effects] Species variability and connectivity in the deep sea: evaluating effects of spatial heterogeneity and hydrodynamic effects Supplementary material for [L Lins], [2016], [Species variability and connectivity in the deep sea: evaluating effects of spatial heterogeneity and hydrodynamic effects] Supplementary material for [L Lins], [2016], [Species variability and connectivity in the deep sea: evaluating effects of spatial heterogeneity and hydrodynamic effects] Supplementary Figure 1: Partial-18S rDNA phylogeny of Nematoda: Chromadorea. The inferred relationships support a broad taxonomic representation of nematodes in samples from lower shelf and upper slope at the West-Iberian Margin and furthermore indicate neither geographic nor depth clustering between ‘deep’ and ‘shallow’ taxa at any level of the tree topology. Reconstruction of nematode 18S relationships was conducted using Maximum Likelihood. Bootstrap support values were generated using 1000 replicates and are presented as node support. The analyses were performed by means of Randomized Axelerated Maximum Likelihood (RAxML). Branch (line) width represents statistical support. Sequences retrieved from Genbank are represented by their Genbank Accession numbers. Orders and Families are annotated as branch labels. PERMANOVA table of results (2-factor design) Source df SS MS Pseudo-F P(perm) Unique perms Depth 1 105.29 -

World Database of Free-Living Marine Nematodes Editor Workshop Report
World Database of Free-Living Marine Nematodes Editor Workshop Ghent University – Marine Biology Research Group – Ghent, Belgium 05 - 07 September 2018 Email to contact all Nemys editors: [email protected] Report Attendants Editors: Tania Nara Bezerra, Ann Vanreusel, Mike Hodda, Zeng Zhao, Ursula Eisendle-Flöckner, Oleksandr Holovachov, Virág Venekey, Nic Smol, Wilfrida Decraemer, Dmitry Miljutin, Vadim Mokievsky Excused: Daniel Leduc, Jyotsna Sharma, Reyes Peña Santiago, Alexei Tchesunov WoRMS Data Management Team (DMT): Bart Vanhoorne, Wim Decock, Thomas Lanssens, Ricardo Simões Excused: Leen Vandepitte The first Nemys Editors Workshop in February 2015 result in a considerable improvement of the database and in 2016 during the Meiofauna Conference in Crete, where some of the editors were present, some issues were also discussed. These possibilities of having the editors working alongside provide an efficient work in a shorter time. We definitely need to have these workshops periodically. The objectives of this workshop were: (i) to evaluate the work that has been done over the past three years and the maintenance of the database, (ii) to welcome the editors of terrestrial and fresh water nematodes, set clear goals, assign tasks and initiate the practical work related to new initiatives and (iii) to outline practices for outreach and scientific output for Nemys (e.g. scientific papers, presentations of the progress, plans and results at an upcoming conference). On the first day it was asked to have a rapporteur for each session in order to have it registered. The original minutes are on a separate document and were used to generate this report. This report includes: I. -
Free-Living Marine Nematodes from San Antonio Bay (Río Negro, Argentina)
A peer-reviewed open-access journal ZooKeys 574: 43–55Free-living (2016) marine nematodes from San Antonio Bay (Río Negro, Argentina) 43 doi: 10.3897/zookeys.574.7222 DATA PAPER http://zookeys.pensoft.net Launched to accelerate biodiversity research Free-living marine nematodes from San Antonio Bay (Río Negro, Argentina) Gabriela Villares1, Virginia Lo Russo1, Catalina Pastor de Ward1, Viviana Milano2, Lidia Miyashiro3, Renato Mazzanti3 1 Laboratorio de Meiobentos LAMEIMA-CENPAT-CONICET, Boulevard Brown 2915, U9120ACF, Puerto Madryn, Argentina 2 Universidad Nacional de la Patagonia San Juan Bosco, sede Puerto Madryn. Boulevard Brown 3051, U9120ACF, Puerto Madryn, Argentina 3Centro de Cómputos CENPAT-CONICET, Boulevard Brown 2915, U9120ACF, Puerto Madryn, Argentina Corresponding author: Gabriela Villares ([email protected]) Academic editor: H-P Fagerholm | Received 18 November 2015 | Accepted 11 February 2016 | Published 28 March 2016 http://zoobank.org/3E8B6DD5-51FA-499D-AA94-6D426D5B1913 Citation: Villares G, Lo Russo V, Pastor de Ward C, Milano V, Miyashiro L, Mazzanti R (2016) Free-living marine nematodes from San Antonio Bay (Río Negro, Argentina). ZooKeys 574: 43–55. doi: 10.3897/zookeys.574.7222 Abstract The dataset of free-living marine nematodes of San Antonio Bay is based on sediment samples collected in February 2009 during doctoral theses funded by CONICET grants. A total of 36 samples has been taken at three locations in the San Antonio Bay, Santa Cruz Province, Argentina on the coastal littoral at three tidal levels. This presents a unique and important collection for benthic biodiversity assessment of Patagonian nematodes as this area remains one of the least known regions. -

Sensitivity of Ribosomal RNA Character Sampling in the Phylogeny of Rhabditida
Journal of Nematology 47(4):337–355. 2015. Ó The Society of Nematologists 2015. Sensitivity of Ribosomal RNA Character Sampling in the Phylogeny of Rhabditida 1 2 2 OLEKSANDR HOLOVACHOV, LAUREN CAMP, AND STEVEN A. NADLER Abstract: Near-full-length 18S and 28S rRNA gene sequences were obtained for 33 nematode species. Datasets were constructed based on secondary structure and progressive multiple alignments, and clades were compared for phylogenies inferred by Bayesian and maximum likelihood methods. Clade comparisons were also made following removal of ambiguously aligned sites as determined using the program ProAlign. Different alignments of these data produced tree topologies that differed, sometimes markedly, when analyzed by the same inference method. With one exception, the same alignment produced an identical tree topology when analyzed by different methods. Removal of ambiguously aligned sites altered the tree topology and also reduced resolution. Nematode clades were sensitive to differences in multiple alignments, and more than doubling the amount of sequence data by addition of 28S rRNA did not fully mitigate this result. Although some individual clades showed substantially higher support when 28S data were combined with 18S data, the combined analysis yielded no statistically significant increases in the number of clades receiving higher support when compared to the 18S data alone. Secondary structure alignment increased accuracy in positional homology assignment and, when used in combination with paired-site substitution models, these structural hypotheses of characters and improved models of character state change yielded high levels of phylogenetic resolution. Phylogenetic results included strong support for inclusion of Daubaylia potomaca within Cephalobidae, whereas the position of Fescia grossa within Tylenchina varied depending on the alignment, and the relationships among Rhabditidae, Diplogastridae, and Bunonematidae were not resolved. -

Developmental Plasticity, Ecology, and Evolutionary Radiation of Nematodes of Diplogastridae
Developmental Plasticity, Ecology, and Evolutionary Radiation of Nematodes of Diplogastridae Dissertation der Mathematisch-Naturwissenschaftlichen Fakultät der Eberhard Karls Universität Tübingen zur Erlangung des Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) vorgelegt von Vladislav Susoy aus Berezniki, Russland Tübingen 2015 Gedruckt mit Genehmigung der Mathematisch-Naturwissenschaftlichen Fakultät der Eberhard Karls Universität Tübingen. Tag der mündlichen Qualifikation: 5 November 2015 Dekan: Prof. Dr. Wolfgang Rosenstiel 1. Berichterstatter: Prof. Dr. Ralf J. Sommer 2. Berichterstatter: Prof. Dr. Heinz-R. Köhler 3. Berichterstatter: Prof. Dr. Hinrich Schulenburg Acknowledgements I am deeply appreciative of the many people who have supported my work. First and foremost, I would like to thank my advisors, Professor Ralf J. Sommer and Dr. Matthias Herrmann for giving me the opportunity to pursue various research projects as well as for their insightful scientific advice, support, and encouragement. I am also very grateful to Matthias for introducing me to nematology and for doing an excellent job of organizing fieldwork in Germany, Arizona and on La Réunion. I would like to thank the members of my examination committee: Professor Heinz-R. Köhler and Professor Hinrich Schulenburg for evaluating this dissertation and Dr. Felicity Jones, Professor Karl Forchhammer, and Professor Rolf Reuter for being my examiners. I consider myself fortunate for having had Dr. Erik J. Ragsdale as a colleague for several years, and more than that to count him as a friend. We have had exciting collaborations and great discussions and I would like to thank you, Erik, for your attention, inspiration, and thoughtful feedback. I also want to thank Erik and Orlando de Lange for reading over drafts of this dissertation and spelling out some nuances of English writing. -

Ahead of Print Online Version Phylogenetic Relationships of Some
Ahead of print online version FOLIA PARASITOLOGICA 58[2]: 135–148, 2011 © Institute of Parasitology, Biology Centre ASCR ISSN 0015-5683 (print), ISSN 1803-6465 (online) http://www.paru.cas.cz/folia/ Phylogenetic relationships of some spirurine nematodes (Nematoda: Chromadorea: Rhabditida: Spirurina) parasitic in fishes inferred from SSU rRNA gene sequences Eva Černotíková1,2, Aleš Horák1 and František Moravec1 1 Institute of Parasitology, Biology Centre of the Academy of Sciences of the Czech Republic, Branišovská 31, 370 05 České Budějovice, Czech Republic; 2 Faculty of Science, University of South Bohemia, Branišovská 31, 370 05 České Budějovice, Czech Republic Abstract: Small subunit rRNA sequences were obtained from 38 representatives mainly of the nematode orders Spirurida (Camalla- nidae, Cystidicolidae, Daniconematidae, Philometridae, Physalopteridae, Rhabdochonidae, Skrjabillanidae) and, in part, Ascaridida (Anisakidae, Cucullanidae, Quimperiidae). The examined nematodes are predominantly parasites of fishes. Their analyses provided well-supported trees allowing the study of phylogenetic relationships among some spirurine nematodes. The present results support the placement of Cucullanidae at the base of the suborder Spirurina and, based on the position of the genus Philonema (subfamily Philoneminae) forming a sister group to Skrjabillanidae (thus Philoneminae should be elevated to Philonemidae), the paraphyly of the Philometridae. Comparison of a large number of sequences of representatives of the latter family supports the paraphyly of the genera Philometra, Philometroides and Dentiphilometra. The validity of the newly included genera Afrophilometra and Carangi- nema is not supported. These results indicate geographical isolation has not been the cause of speciation in this parasite group and no coevolution with fish hosts is apparent. On the contrary, the group of South-American species ofAlinema , Nilonema and Rumai is placed in an independent branch, thus markedly separated from other family members. -

Nematode Taxonomic Diversity and Community Structure: Indicators of Environmental Conditions at Keetham Lake, Agra
NEMATODE TAXONOMIC DIVERSITY AND COMMUNITY STRUCTURE: INDICATORS OF ENVIRONMENTAL CONDITIONS AT KEETHAM LAKE, AGRA THESIS SUBMITTED FOR THE AWARD OF THE DEGREE OF DOCTOR OF PHILOSOPHY IN ZOOLOGY BY MALKA MUSTAQIM UNDER THE SUPERVISION OF PROF. QUDSIA TAHSEEN DEPARTMENT OF ZOOLOGY ALIGARH MUSLIM UNIVERSITY ALIGARH-202002 (INDIA) 2017 1 Dedicated to my Beloved Parents and Brothers 2 Qudsia Tahseen, Professor Department of Zoology, PhD, FASc, FNASc Aligarh Muslim University, Aligarh-202002, India Tel: +91 9319624196 E-mail: [email protected] Certificate This is to certify that the entire work presented in the thesis entitled, ‘‘Nematode taxonomic diversity and community structure: indicators of environmental conditions at Keetham Lake, Agra’’ by Ms. Malka Mustaqim is original and was carried out under my supervision. I have permitted Ms. Mustaqim to submit the thesis to Aligarh Muslim University, Aligarh for the award of degree of Doctor of Philosophy in Zoology. (Qudsia Tahseen) Supervisor 3 ANNEXURE-Ι CANDIDATE’S DECLARATION I, Malka Mustaqim, Department of Zoology, certify that the work embodied in this Ph.D. thesis is my own bonafide work carried out by me under the supervision of Prof. Qudsia Tahseen at Aligarh Muslim University, Aligarh. The matter embodied in this Ph.D. thesis has not been submitted for the award of any other degree. I declare that I have faithfully acknowledged, given credit to and referred to the research workers wherever their works have been cited in the text and the body of the thesis. I further certify that I have not willfully lifted up some others work, para, text, data, results, etc. -

(Stsm) Scientific Report
SHORT TERM SCIENTIFIC MISSION (STSM) SCIENTIFIC REPORT This report is submitted for approval by the STSM applicant to the STSM coordinator Action number: CA15219-45333 STSM title: Free-living marine nematodes from the eastern Mediterranean deep sea - connecting COI and 18S rRNA barcodes to structure and function STSM start and end date: 06/02/2020 to 18/3/2020 (short than the planned two months due to the Co-Vid 19 virus pandemic) Grantee name: Zoya Garbuzov PURPOSE OF THE STSM: My Ph.D. thesis is devoted to the population ecology of free-living nematodes inhabiting deep-sea soft substrates of the Mediterranean Levantine Basin. The success of the study largely depends on my ability to accurately identify collected nematodes at the species level, essential for appropriate environmental analysis. Morphological identification of nematodes at the species level is fraught with difficulties, mainly because of their relatively simple body shape and the absence of distinctive morphological characters. Therefore, a combination of morphological identification to genus level and the use of molecular markers to reach species identification is assumed to provide a better distinction of species in this difficult to identify group. My STSM host, Dr. Nikolaos Lampadariou, is an experienced taxonomist and nematode ecologist. In addition, I will have access to the molecular laboratory of Dr. Panagiotis Kasapidis. Both researchers are based at the Hellenic Center for Marine Research (HCMR) in Crete and this STSM is aimed at combining morphological taxonomy, under the supervision of Dr. Lampadariou, with my recently acquired experience in nematode molecular taxonomy for relating molecular identifiers to nematode morphology. -
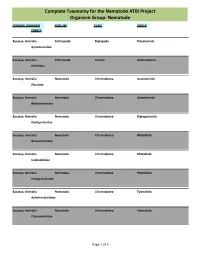
Complete Taxonomy for the Nematode ATBI Project Organism Group: Nematode
Complete Taxonomy for the Nematode ATBI Project Organism Group: Nematode DOMAIN, KINGDOM PHYLUM CLASS ORDER FAMILY Eucarya, Animalia Arthropoda Diplopoda Polydesmida Xystodesmidae Eucarya, Animalia Arthropoda Insecta Hymenoptera Halictidae Eucarya, Animalia Nematoda Chromadorea Araeolaimida Plectidae Eucarya, Animalia Nematoda Chromadorea Araeolaimida Rhabdolaimidae Eucarya, Animalia Nematoda Chromadorea Diplogasterida Diplogasteridae Eucarya, Animalia Nematoda Chromadorea Rhabditida Bunonematidae Eucarya, Animalia Nematoda Chromadorea Rhabditida Cephalobidae Eucarya, Animalia Nematoda Chromadorea Rhabditida Panagrolaimidae Eucarya, Animalia Nematoda Chromadorea Tylenchida Aphelenchoididae Eucarya, Animalia Nematoda Chromadorea Tylenchida Criconematidae Page 1 of 4 Complete Taxonomy for the Nematode ATBI Project Organism Group: Nematode DOMAIN, KINGDOM PHYLUM CLASS ORDER FAMILY Eucarya, Animalia Nematoda Chromadorea Tylenchida Hoplolaimidae Eucarya, Animalia Nematoda Enoplea Dorylaimida Actinolaimidae Eucarya, Animalia Nematoda Enoplea Dorylaimida Aporcelaimidae Eucarya, Animalia Nematoda Enoplea Dorylaimida Dorylaimidae Eucarya, Animalia Nematoda Enoplea Dorylaimida Longidoridae Eucarya, Animalia Nematoda Enoplea Dorylaimida Nygolaimidae Eucarya, Animalia Nematoda Enoplea Dorylaimida Qudsianematidae Eucarya, Animalia Nematoda Enoplea Enoplida Alaimidae Eucarya, Animalia Nematoda Enoplea Enoplida Ironidae Eucarya, Animalia Nematoda Enoplea Enoplida Prismatolaimidae Page 2 of 4 Complete Taxonomy for the Nematode ATBI Project Organism Group: -

Succession of Nematodes During Composting Processess and Their Potential As Indicators of Compost Maturity
Succession of nematodes during composting processess and their potential as indicators of compost maturity Hanne Steel Promoters: Prof. dr. Wim Bert (UGent) Prof. dr. Tom Moens (UGent) Thesis submitted to obtain the degree of doctor in Sciences, Biology Proefschrift voorgelegd tot het bekomen van de graad van doctor in de Wetenschappen, Biologie Dit werk werd mogelijk gemaakt door een beurs van het Fonds Wetenschappelijk Onderzoek- Vlaanderen (FWO) This work was supported by a grant of the Foundation for Scientific Research, Flanders (FWO) 3 Reading Committee: Prof. dr. Deborah Neher (University of Vermont, USA) Dr. Thomaé Kakouli-Duarte (Institute of Technology Carlow, Ireland) Prof. dr. Magda Vincx (Ghent University, Belgium) Dr. Eduardo de la Peña (Ghent University, Belgium) Examination Committee: Prof. dr. Koen Sabbe (chairman, Ghent University, Belgium) Prof. dr. Wim Bert (secretary, promotor, Ghent University, Belgium) Prof. dr. Tom Moens (promotor, Ghent University, Belgium) Prof. dr. Deborah Neher (University of Vermont, USA) Dr. Thomaé Kakouli-Duarte (Institute of Technology Carlow, Ireland) Prof. dr. Magda Vincx (Ghent University, Belgium) Prof. dr. Wilfrida Decraemer (Royal Belgian Institute of Natural Sciences, Belgium) Dr. Eduardo de la Peña (Ghent University, Belgium) Dr. Ir. Bart Vandecasteele (Institute for Agricultural and Fisheries Research, Belgium) 5 Acknowledgments Eindelijk is het zover! Ik mag mijn dankwoord schrijven, iets waar ik stiekem al heel lang naar uitkijk en dat alleen maar kan betekenen dat mijn doctoraat bijna klaar is. JOEPIE! De voorbije 5 jaar waren zonder twijfel leuk, leerrijk en ontzettend boeiend. Maar… jawel doctoreren is ook een project van lange adem, met vallen en opstaan, met zin en tegenzin, met geluk en tegenslag, met fantastische hoogtes maar soms ook laagtes….Nu ik er zo over nadenk en om in een vertrouwd thema te blijven: doctoreren verschilt eigenlijk niet zo gek veel van een composteringsproces, dat bij voorkeur trouwens ook veel adem (zuurstof) ter beschikking heeft. -

Nematoda, Chromadorida, Selachinematidae) from the West Coast of Sweden and an Updated Phylogeny of Chromadoria
Marine Biodiversity (2020) 50:113 https://doi.org/10.1007/s12526-020-01129-w ORIGINAL PAPER Description of a new marine predatory nematode Latronema dyngi sp. nov. (Nematoda, Chromadorida, Selachinematidae) from the west coast of Sweden and an updated phylogeny of Chromadoria Mohammed Ahmed1 & Oleksandr Holovachov1 Received: 3 July 2020 /Revised: 8 October 2020 /Accepted: 15 October 2020 # The Author(s) 2020 Abstract A new nematode species, Latronema dyngi sp. nov., is described from Skagerrak off the west coast of Sweden with the type locality near Dyngö island. Latronema dyngi sp. nov. is characterized by multispiral amphideal fovea with circular outline, 0.2– 0.3 corresponding body diameters wide in males and 0.1–0.2 corresponding body diameters wide in females, 12 cuticular longitudinal ridges and 18–27 precloacal supplements in males. Latronema dyngi sp. nov. most closely resembles L. orcinum in terms of body length; demanian ratios a, b, c and c′; number of amphid turns in males; and the ratio of spicule length to cloacal body diameter. The two species can be differentiated by the number longitudinal ridges on the cuticle (12 for Latronema dyngi sp. nov. vs 20–22 for L. orcinum) and spicule length (65–78 μmforL. dyngi vs 60 for L. orcinum) and shape (weakly arcuate for L. dyngi sp. nov. vs strongly arcuate for L. orcinum). We also performed a maximum likelihood phylogenetic analysis on over 250 nematodes of the subclass Chromadoria based on their nearly full-length 18S rDNA sequences. In agreement with previous studies, our analysis supported Selachinematidae as a monophyletic group and placed Richtersia Steiner, 1916 within Desmodoridae Filipjev, 1922 or just outside of the main Desmodorida clade with the latter placement not well supported. -

Taxonomy Assignment Approach Determines the Efficiency of Identification of Metabarcodes in Marine Nematodes
Taxonomy assignment approach determines the efficiency of identification of metabarcodes in marine nematodes Oleksandr Holovachov1, Quiterie Haenel2, Sarah J. Bourlat3 and Ulf Jondelius1 1Department of Zoology, Swedish Museum of Natural History, Stockholm, Sweden 2Zoological Institute, University of Basel, Switzerland 3Department of Marine Sciences, University of Gothenburg, Sweden Abstract: Precision and reliability of barcode-based biodiversity assessment can be affected at several steps during acquisition and analysis of the data. Identification of barcodes is one of the crucial steps in the process and can be accomplished using several different approaches, namely, alignment-based, probabilistic, tree-based and phylogeny-based. Number of identified sequences in the reference databases affects the precision of identification. This paper compares the identification of marine nematode barcodes using alignment-based, tree-based and phylogeny-based approaches. Because the nematode reference dataset is limited in its taxonomic scope, barcodes can only be assigned to higher taxonomic categories, families. Phylogeny-based approach using Evolutionary Placement Algorithm provided the largest number of positively assigned metabarcodes and was least affected by erroneous sequences and limitations of reference data, comparing to alignment- based and tree-based approaches. Key words: biodiversity, identification, barcode, nematodes, meiobenthos. 1 1. Introduction Metabarcoding studies based on high throughput sequencing of amplicons from marine samples have reshaped our understanding of the biodiversity of marine microscopic eukaryotes, revealing a much higher diversity than previously known [1]. Early metabarcoding of the slightly larger sediment-dwelling meiofauna have mainly focused on scoring relative diversity of taxonomic groups [1-3]. The next step in metabarcoding: identification of species, is limited by the available reference database, which is sparse for most marine taxa, and by the matching algorithms.