Sensitivity of Ribosomal RNA Character Sampling in the Phylogeny of Rhabditida
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Monophyly of Clade III Nematodes Is Not Supported by Phylogenetic Analysis of Complete Mitochondrial Genome Sequences
UC Davis UC Davis Previously Published Works Title Monophyly of clade III nematodes is not supported by phylogenetic analysis of complete mitochondrial genome sequences Permalink https://escholarship.org/uc/item/7509r5vp Journal BMC Genomics, 12(1) ISSN 1471-2164 Authors Park, Joong-Ki Sultana, Tahera Lee, Sang-Hwa et al. Publication Date 2011-08-03 DOI http://dx.doi.org/10.1186/1471-2164-12-392 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Park et al. BMC Genomics 2011, 12:392 http://www.biomedcentral.com/1471-2164/12/392 RESEARCHARTICLE Open Access Monophyly of clade III nematodes is not supported by phylogenetic analysis of complete mitochondrial genome sequences Joong-Ki Park1*, Tahera Sultana2, Sang-Hwa Lee3, Seokha Kang4, Hyong Kyu Kim5, Gi-Sik Min2, Keeseon S Eom6 and Steven A Nadler7 Abstract Background: The orders Ascaridida, Oxyurida, and Spirurida represent major components of zooparasitic nematode diversity, including many species of veterinary and medical importance. Phylum-wide nematode phylogenetic hypotheses have mainly been based on nuclear rDNA sequences, but more recently complete mitochondrial (mtDNA) gene sequences have provided another source of molecular information to evaluate relationships. Although there is much agreement between nuclear rDNA and mtDNA phylogenies, relationships among certain major clades are different. In this study we report that mtDNA sequences do not support the monophyly of Ascaridida, Oxyurida and Spirurida (clade III) in contrast to results for nuclear rDNA. Results from mtDNA genomes show promise as an additional independently evolving genome for developing phylogenetic hypotheses for nematodes, although substantially increased taxon sampling is needed for enhanced comparative value with nuclear rDNA. -

Testing the Hypothesis of Recent Population Expansions in Nematode Parasites of Human-Associated Hosts
Heredity (2005) 94, 426–434 & 2005 Nature Publishing Group All rights reserved 0018-067X/05 $30.00 www.nature.com/hdy Testing the hypothesis of recent population expansions in nematode parasites of human-associated hosts DA Morrison and J Ho¨glund Department of Parasitology (SWEPAR), National Veterinary Institute and Swedish University of Agricultural Sciences, 751 89 Uppsala, Sweden It has been predicted that parasites of human-associated prediction. However, it is likely that the situation is more organisms (eg humans, domestic pets, farm animals, complicated than the simple hypothesis test suggests, and agricultural and silvicultural plants) are more likely to show those species that do not fit the predicted general pattern rapid recent population expansions than are parasites of provide interesting insights into other evolutionary processes other hosts. Here, we directly test the generality of this that influence the historical population genetics of host– demographic prediction for species of parasitic nematodes parasite relationships. These processes include the effects of that currently have mitochondrial sequence data available in postglacial migrations, evolutionary relationships and possi- the literature or the public-access genetic databases. Of the bly life-history characteristics. Furthermore, the analysis 23 host/parasite combinations analysed, there are seven highlights the limitations of this form of bioinformatic data- human-associated parasite species with expanding popula- mining, in comparison to controlled experimental -

Developmental Plasticity, Ecology, and Evolutionary Radiation of Nematodes of Diplogastridae
Developmental Plasticity, Ecology, and Evolutionary Radiation of Nematodes of Diplogastridae Dissertation der Mathematisch-Naturwissenschaftlichen Fakultät der Eberhard Karls Universität Tübingen zur Erlangung des Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) vorgelegt von Vladislav Susoy aus Berezniki, Russland Tübingen 2015 Gedruckt mit Genehmigung der Mathematisch-Naturwissenschaftlichen Fakultät der Eberhard Karls Universität Tübingen. Tag der mündlichen Qualifikation: 5 November 2015 Dekan: Prof. Dr. Wolfgang Rosenstiel 1. Berichterstatter: Prof. Dr. Ralf J. Sommer 2. Berichterstatter: Prof. Dr. Heinz-R. Köhler 3. Berichterstatter: Prof. Dr. Hinrich Schulenburg Acknowledgements I am deeply appreciative of the many people who have supported my work. First and foremost, I would like to thank my advisors, Professor Ralf J. Sommer and Dr. Matthias Herrmann for giving me the opportunity to pursue various research projects as well as for their insightful scientific advice, support, and encouragement. I am also very grateful to Matthias for introducing me to nematology and for doing an excellent job of organizing fieldwork in Germany, Arizona and on La Réunion. I would like to thank the members of my examination committee: Professor Heinz-R. Köhler and Professor Hinrich Schulenburg for evaluating this dissertation and Dr. Felicity Jones, Professor Karl Forchhammer, and Professor Rolf Reuter for being my examiners. I consider myself fortunate for having had Dr. Erik J. Ragsdale as a colleague for several years, and more than that to count him as a friend. We have had exciting collaborations and great discussions and I would like to thank you, Erik, for your attention, inspiration, and thoughtful feedback. I also want to thank Erik and Orlando de Lange for reading over drafts of this dissertation and spelling out some nuances of English writing. -

Ahead of Print Online Version Phylogenetic Relationships of Some
Ahead of print online version FOLIA PARASITOLOGICA 58[2]: 135–148, 2011 © Institute of Parasitology, Biology Centre ASCR ISSN 0015-5683 (print), ISSN 1803-6465 (online) http://www.paru.cas.cz/folia/ Phylogenetic relationships of some spirurine nematodes (Nematoda: Chromadorea: Rhabditida: Spirurina) parasitic in fishes inferred from SSU rRNA gene sequences Eva Černotíková1,2, Aleš Horák1 and František Moravec1 1 Institute of Parasitology, Biology Centre of the Academy of Sciences of the Czech Republic, Branišovská 31, 370 05 České Budějovice, Czech Republic; 2 Faculty of Science, University of South Bohemia, Branišovská 31, 370 05 České Budějovice, Czech Republic Abstract: Small subunit rRNA sequences were obtained from 38 representatives mainly of the nematode orders Spirurida (Camalla- nidae, Cystidicolidae, Daniconematidae, Philometridae, Physalopteridae, Rhabdochonidae, Skrjabillanidae) and, in part, Ascaridida (Anisakidae, Cucullanidae, Quimperiidae). The examined nematodes are predominantly parasites of fishes. Their analyses provided well-supported trees allowing the study of phylogenetic relationships among some spirurine nematodes. The present results support the placement of Cucullanidae at the base of the suborder Spirurina and, based on the position of the genus Philonema (subfamily Philoneminae) forming a sister group to Skrjabillanidae (thus Philoneminae should be elevated to Philonemidae), the paraphyly of the Philometridae. Comparison of a large number of sequences of representatives of the latter family supports the paraphyly of the genera Philometra, Philometroides and Dentiphilometra. The validity of the newly included genera Afrophilometra and Carangi- nema is not supported. These results indicate geographical isolation has not been the cause of speciation in this parasite group and no coevolution with fish hosts is apparent. On the contrary, the group of South-American species ofAlinema , Nilonema and Rumai is placed in an independent branch, thus markedly separated from other family members. -

Nematode Taxonomic Diversity and Community Structure: Indicators of Environmental Conditions at Keetham Lake, Agra
NEMATODE TAXONOMIC DIVERSITY AND COMMUNITY STRUCTURE: INDICATORS OF ENVIRONMENTAL CONDITIONS AT KEETHAM LAKE, AGRA THESIS SUBMITTED FOR THE AWARD OF THE DEGREE OF DOCTOR OF PHILOSOPHY IN ZOOLOGY BY MALKA MUSTAQIM UNDER THE SUPERVISION OF PROF. QUDSIA TAHSEEN DEPARTMENT OF ZOOLOGY ALIGARH MUSLIM UNIVERSITY ALIGARH-202002 (INDIA) 2017 1 Dedicated to my Beloved Parents and Brothers 2 Qudsia Tahseen, Professor Department of Zoology, PhD, FASc, FNASc Aligarh Muslim University, Aligarh-202002, India Tel: +91 9319624196 E-mail: [email protected] Certificate This is to certify that the entire work presented in the thesis entitled, ‘‘Nematode taxonomic diversity and community structure: indicators of environmental conditions at Keetham Lake, Agra’’ by Ms. Malka Mustaqim is original and was carried out under my supervision. I have permitted Ms. Mustaqim to submit the thesis to Aligarh Muslim University, Aligarh for the award of degree of Doctor of Philosophy in Zoology. (Qudsia Tahseen) Supervisor 3 ANNEXURE-Ι CANDIDATE’S DECLARATION I, Malka Mustaqim, Department of Zoology, certify that the work embodied in this Ph.D. thesis is my own bonafide work carried out by me under the supervision of Prof. Qudsia Tahseen at Aligarh Muslim University, Aligarh. The matter embodied in this Ph.D. thesis has not been submitted for the award of any other degree. I declare that I have faithfully acknowledged, given credit to and referred to the research workers wherever their works have been cited in the text and the body of the thesis. I further certify that I have not willfully lifted up some others work, para, text, data, results, etc. -

Strongyloides Stercoralis 1
BIO 475 - Parasitology Spring 2009 Stephen M. Shuster Northern Arizona University http://www4.nau.edu/isopod Lecture 19 Order Trichurida b. Trichinella spiralis 1. omnivore parasite 2. larvae encyst in muscle, cause "trichinosis" Life Cycle: Unusual Aspects a. Adults inhabit intestine, females embed in intestinal wall. b. Eggs mature in female uterus, larvae (J1) enter blood and lymph, travel to vascularized muscle. c. Larvae encyst in muscle, wait to be eaten. d. J4 hatch and mature into adults. 1 2 Mozart may have died from eating undercooked pork Mozart's death in 1791 at age 35 may have been from trichinosis, according to a review of historical documents and examination of other theories. Medical researcher Jan Hirschmann's analysis of the composer's final illness appears in the June 11 Archives of Internal Medicine. Hirschmann is a UW professor of medicine in the Division of General Internal Medicine and practices at Veterans Affairs Puget Sound Health Care System. Hirschmann's study of Mozart's symptoms and the circumstances surrounding his death includes a letter in which the composer mentions eating pork cutlets. The letter, written 44 days before he died, correlates with the incubation period for trichinosis. The nature of Mozart's fatal illness has vexed historians and led to much speculation, because no bodily remains are left for evidence. The official but vague cause of Mozart's death was listed as "severe military fever". The symptoms his family described included edema without dyspnea, limb pain and swelling. Mozart composed music and communicated until his last moments. Order Trichurida Capillaria spp. -
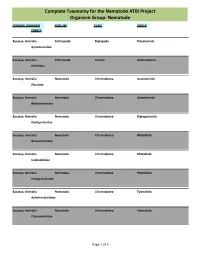
Complete Taxonomy for the Nematode ATBI Project Organism Group: Nematode
Complete Taxonomy for the Nematode ATBI Project Organism Group: Nematode DOMAIN, KINGDOM PHYLUM CLASS ORDER FAMILY Eucarya, Animalia Arthropoda Diplopoda Polydesmida Xystodesmidae Eucarya, Animalia Arthropoda Insecta Hymenoptera Halictidae Eucarya, Animalia Nematoda Chromadorea Araeolaimida Plectidae Eucarya, Animalia Nematoda Chromadorea Araeolaimida Rhabdolaimidae Eucarya, Animalia Nematoda Chromadorea Diplogasterida Diplogasteridae Eucarya, Animalia Nematoda Chromadorea Rhabditida Bunonematidae Eucarya, Animalia Nematoda Chromadorea Rhabditida Cephalobidae Eucarya, Animalia Nematoda Chromadorea Rhabditida Panagrolaimidae Eucarya, Animalia Nematoda Chromadorea Tylenchida Aphelenchoididae Eucarya, Animalia Nematoda Chromadorea Tylenchida Criconematidae Page 1 of 4 Complete Taxonomy for the Nematode ATBI Project Organism Group: Nematode DOMAIN, KINGDOM PHYLUM CLASS ORDER FAMILY Eucarya, Animalia Nematoda Chromadorea Tylenchida Hoplolaimidae Eucarya, Animalia Nematoda Enoplea Dorylaimida Actinolaimidae Eucarya, Animalia Nematoda Enoplea Dorylaimida Aporcelaimidae Eucarya, Animalia Nematoda Enoplea Dorylaimida Dorylaimidae Eucarya, Animalia Nematoda Enoplea Dorylaimida Longidoridae Eucarya, Animalia Nematoda Enoplea Dorylaimida Nygolaimidae Eucarya, Animalia Nematoda Enoplea Dorylaimida Qudsianematidae Eucarya, Animalia Nematoda Enoplea Enoplida Alaimidae Eucarya, Animalia Nematoda Enoplea Enoplida Ironidae Eucarya, Animalia Nematoda Enoplea Enoplida Prismatolaimidae Page 2 of 4 Complete Taxonomy for the Nematode ATBI Project Organism Group: -

Succession of Nematodes During Composting Processess and Their Potential As Indicators of Compost Maturity
Succession of nematodes during composting processess and their potential as indicators of compost maturity Hanne Steel Promoters: Prof. dr. Wim Bert (UGent) Prof. dr. Tom Moens (UGent) Thesis submitted to obtain the degree of doctor in Sciences, Biology Proefschrift voorgelegd tot het bekomen van de graad van doctor in de Wetenschappen, Biologie Dit werk werd mogelijk gemaakt door een beurs van het Fonds Wetenschappelijk Onderzoek- Vlaanderen (FWO) This work was supported by a grant of the Foundation for Scientific Research, Flanders (FWO) 3 Reading Committee: Prof. dr. Deborah Neher (University of Vermont, USA) Dr. Thomaé Kakouli-Duarte (Institute of Technology Carlow, Ireland) Prof. dr. Magda Vincx (Ghent University, Belgium) Dr. Eduardo de la Peña (Ghent University, Belgium) Examination Committee: Prof. dr. Koen Sabbe (chairman, Ghent University, Belgium) Prof. dr. Wim Bert (secretary, promotor, Ghent University, Belgium) Prof. dr. Tom Moens (promotor, Ghent University, Belgium) Prof. dr. Deborah Neher (University of Vermont, USA) Dr. Thomaé Kakouli-Duarte (Institute of Technology Carlow, Ireland) Prof. dr. Magda Vincx (Ghent University, Belgium) Prof. dr. Wilfrida Decraemer (Royal Belgian Institute of Natural Sciences, Belgium) Dr. Eduardo de la Peña (Ghent University, Belgium) Dr. Ir. Bart Vandecasteele (Institute for Agricultural and Fisheries Research, Belgium) 5 Acknowledgments Eindelijk is het zover! Ik mag mijn dankwoord schrijven, iets waar ik stiekem al heel lang naar uitkijk en dat alleen maar kan betekenen dat mijn doctoraat bijna klaar is. JOEPIE! De voorbije 5 jaar waren zonder twijfel leuk, leerrijk en ontzettend boeiend. Maar… jawel doctoreren is ook een project van lange adem, met vallen en opstaan, met zin en tegenzin, met geluk en tegenslag, met fantastische hoogtes maar soms ook laagtes….Nu ik er zo over nadenk en om in een vertrouwd thema te blijven: doctoreren verschilt eigenlijk niet zo gek veel van een composteringsproces, dat bij voorkeur trouwens ook veel adem (zuurstof) ter beschikking heeft. -

Ancylostoma Duodenale, Necator Americanus, Human-Specific Hookworms: Life Cycle, Health Implications at Metapathogen
http://www.MetaPathogen.com: Human-specific hookworms Note: description of Ancylostoma caninum (dog hookworm) can be found here. ● Ancylostoma duodenale taxonomy ● Necator americanus taxonomy ● Brief facts ● Developmental stages ● References Ancylostoma duodenale taxonomy cellular organisms - Eukaryota - Fungi/Metazoa group - Metazoa - Eumetazoa - Bilateria - Pseudocoelomata - Nematoda - Chromadorea - Rhabditida - Strongylida - Ancylostomatoidea - Ancylostomatidae - Ancylostomatinae - Ancylostoma - Ancylostoma duodenale Necator americanus taxonomy cellular organisms - Eukaryota - Fungi/Metazoa group - Metazoa - Eumetazoa - Bilateria - Pseudocoelomata - Nematoda - Chromadorea - Rhabditida - Strongylida - Ancylostomatoidea - Ancylostomatidae - Bunostominae - Necator - Necator americanus Brief facts ● Together with human roundworms (see Ascaris lumbricoides at MetaPathogen) and whipworms (Trichuris trichiura), hookworms belong to a group of so-called soil-transmitted helminths that represent one of the world's most important causes of physical and intellectual growth retardation. ● Hookworm infection in humans is usually caused by one of two species of nematodes (roundworms) - Necator americanus or Ancylostoma duodenale. N. americanus is most common human-specific hookworm worldwide, distribution of A. duodenale is geographically more restricted. Both N. americanus and A. duodenale are found in Africa, Asia and the Americas. Necator americanus predominates in the Americas and Australia, while only A. duodenale is found in the Middle East, -

Thesis Outline
UNIVERSITY OF SOUTHAMPTON Faculty of Engineering, Science, and Mathematics School of Ocean and Earth Science Resolving phylogenetic relationships within the order Enoplida (Phylum Nematoda) by Holly Marie Bik Thesis of the degree of Doctor of Philosophy January 2010 Graduate School of the National Oceanography Centre, Southampton This PhD Dissertation by Holly Marie Bik has been produced under the supervision of the following persons Supervisors Prof John Lambshead Dr Lawrence Hawkins Dr Alan Hughes Dr David Lunt Research Advisor Prof W. Kelley Thomas Chair of Advisory Panel Dr Martin Sheader 2 UNIVERSITY OF SOUTHAMPTON ABSTRACT FACULTY OF ENGINEERING, SCIENCE & MATHEMATICS SCHOOL OF OCEAN & EARTH SCIENCE Doctor of Philosophy RESOLVING PHYLOGENETIC RELATIONSHIPS WITHIN THE ORDER ENOPLIDA (PHYLUM NEMATODA) by Holly Marie Bik The Order Enoplida (Phylum Nematoda) has been proposed as a divergent nematode lineage—Enoplid nematodes are thought to exhibit morphological and developmental characteristics present in the ‘ancestral nematode’. However, previous molecular phylogenies have failed to unequivocally confirm the position of this group. The Enoplida is primarily comprised of free-living marine species; if these taxa represent close relatives of the nematode ancestor, this relationship would presumably imply a marine origin for the phylum. Prior to this investigation, few publically available gene sequences existed for Enoplid nematodes, and published sequences represented only shallow water fauna from Northwest Europe. This study has aimed to improve resolution at the base of the nematode tree, using drastically increased taxon-sampling within the previously neglected Enoplid clade. Morphological identifications, nuclear gene sequences (18S and 28S rRNA), and mitochondrial gene sequences (Cox1) were obtained from marine specimens representing a variety of deep-sea and intertidal habitats. -

Thesis Final.Indd
Insects on Individual Plants: Plant quality, plant diversity and aboveground-belowground effects Olga Kostenko Thesis committee Promotor Prof. Dr. W. H. Van der Putten Professor of Functional Biodiversity Laboratory of Nematology Wageningen University Co-promotor Dr. T. M. Bezemer Senior scientist Netherlands Institute of Ecology, Wageningen Other members Prof. Dr. D. Bonte, Ghent University, Belgium Prof. Dr. N. M. Van Dam, Radboud University Nijmegen Prof. Dr. L. Brussaard, Wageningen University Dr. E. H. Poelman, Wageningen University This research was conducted under the auspices of the C.T. de Wit Graduate School for Production Ecology & Resource Conservation (PE&RC) Insects on Individual Plants: Plant quality, plant diversity and aboveground-belowground effects Olga Kostenko Thesis submitted in fulfillment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. Dr. M.J. Kropff, in the presence of the Thesis Committee appointed by the Academic Board to be defended in public on Friday 14th February 2014 at 4 p.m. at the Aula. Olga Kostenko Insects on Individual Plants: Plant quality, plant diversity and aboveground- belowground effects, 232 pages. PhD thesis, Wageningen University, Wageningen, NL (2014) With references, with summaries in Dutch and English ISBN 978-94-6173-863-9 To my Mother Contents Abstract 9 Chapter 1 General introduction 11 Chapter 2 Effects of root herbivory on pyrrolizidine 33 alkaloid content and aboveground plant-herbivore- parasitoid interactions -

A Major Qualifying Project Report Submitted to the Faculty of the WORCESTER POLYTECHNIC INSTITUTE in Partial Fulfillment Of
ANALYSIS OF NSY-1 IN C. ELEGANS AND PARASITIC NEMATODES USING BIOINFORMATICS A Major Qualifying Project Report Submitted to the Faculty Of the WORCESTER POLYTECHNIC INSTITUTE In partial fulfillment of the requirements for the Degree of Bachelor of Science By Maria Grandoni Date: Approved Table of Contents List of Figures…………………………………………………………………………………………………………….………ii List of Tables……………………………………………………………………………………………………………………..iii Abstract……………………………………………………………………………………………………………………………..1 Introduction……………………………………………………………………………………………………………………….2 Nematode anatomy……………………………………………………………………………………………….2 Nematode development………………………………………………………………………………………..2 Nematode lifestyles and life cycles………………………………………………………………………..2 Materials and Methods………………………………………………………………………………………………….….7 Results……………………………………………………………………………………………………………………………….9 N terminal description….………………………………….…………………………………………………..16 Catalytic domain description.………………………………………………………………………………..17 C terminal description…………………………………………………………….…………………………….18 Discussion………………………………………………………………………………………………………………………...23 Acknowledgements……………………………………………………………………………………………………………24 References……………………………………………………………………………………………….……………………….24 Supplementary Material………………………………………………………………………………………………..….27 Step-by-step method to Phylogenetic tree…………………………………………………………....27 Coiled-coil domain predictions……………………………………………………………………………….27 i List of Figures Figure 1: A general phylogenetic tree for the phyla nematoda…………………………………………..4 Figure 2: Overview of ASK-1 structure and function……………………………………………………………6