1. Introduction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Educational Research Applications Abebe M, Et Al
Educational Research Applications Abebe M, et al. Educ Res Appl 5: 175. Review Article DOI: 10.29011/2575-7032.100175 Teaching Students Synthesizing Molecules Mimicking an Existing Drug against Covid-19 Moges Abebe1*, Lashan Eloise Knowles1, Bisrat Hailemeskel2 1Department of Biological and Physical Sciences, Saint Augustine University, Raleigh, NC, USA 2Department of Clinical & Administrative Pharmacy Sciences, College of Pharmacy, Howard University, NW Washington, DC, USA *Corresponding author: Moges Abebe, Department of Biological and Physical Sciences, Saint Augustine University, Raleigh, NC 27610, NC, USA Citation: Abebe M, Knowles LE, Hailemeskel B (2020) Teaching Students Synthesizing Molecules Mimicking an Existing Drug against Covid-19. Educ Res Appl 5: 175. DOI: 10.29011/2575-7032.100175 Received Date: 26 May 2020; Accepted Date: 01 June, 2020; Published Date: 06 June, 2020 Abstract End of semester organic chemistry course projects are valuable learning assessment tools while giving students a creative opportunity and sparking interest for further research investigations. The purpose of this year’s project was to teach students how to synthesize a molecule that potentially mimics an existing drug that works against the COVID-19. The available drugs chosen for the project are those that are proposed to work either by prohibiting the easy entry of the virus into respiratory tissues or those who deprive the virus’s ability to reproduce once they enter the cell. An investigative search in historical literature and the current conditions of the virus enabled students to create a unique and innovative product that requires a cumulative learned knowledge. History has shown that when a new virus becomes pandemic it takes time for researchers to create a drug, test the results, and gets approved by the Food and Drug Administration (FDA) for public availability. -

In This Handout, All of Our Functional Groups Are Presented As Condensed Line Formulas, 2D and 3D Formulas and with Nomenclature Prefixes and Suffixes (If Present)
In this handout, all of our functional groups are presented as condensed line formulas, 2D and 3D formulas and with nomenclature prefixes and suffixes (if present). Organic names are built on a foundation of alkanes, alkenes and alkynes. Those examples are presented first and you need to know those rules. The strategies can be found in Chapter 4 of our textbook (alkanes: pages 93-98, cycloalkanes 102-104, alkenes: pages 104-110, alkynes: pages 112-113 and combinations of all of them 113-115). After introducing examples of alkanes, alkenes, alkynes and combinations of them, the functional groups are presented in order of priority. A few nomenclature examples are provided for each of the functional groups. Examples of the various functional groups are presented on pages 115-135 in the textbook. Two overview pages are on pages 136-137. Some functional groups have a suffix name when they are the highest priority functional group and a prefix name when they are not the highest priority group, and these are added to the skeletal names with identifying numbers and stereochemistry terms (E and Z for alkenes, R and S for chiral centers and cis and trans for rings). Several low priority functional groups only have a prefix name. A few additional special patterns are shown on pages 98-102. The only way to learn this topic is practice (over and over). The best practice approach is to actually write out the names (on an extra piece of paper or on a white board, and then do it again). The same functional groups are used throughout the entire course. -

Chapter 16: Carboxylic Acids, Esters, and Other Acid Derivatives
Chapter 16: Carboxylic Acids, Esters, and Other Acid Derivatives In Chapter 15, we discussed the carbonyl group and two families of compounds -aldehydes and ketones—that contain C=O group. In this chapter, we discuss four more families of compounds in which the carbonyl group is present: a) carboxylic acid, b) esters, c) amides, d) acid chlorides, and e) acid anhydrides and f) carboxylic acid salts. 16.1 Structure of Carboxylic Acids and Their Derivatives A carboxylic acid is an organic compound whose functional group is the carboxyl group. What is a carboxyl group? A carboxyl group is a carbonyl group (C=O) with a hydroxyl group (—OH) bonded to the carboxyl carbon atom. A general structural representation fit a carboxyl group is Abbreviated linear designations for the carboxyl group are Although we see within a carboxyl group both a carbonyl group (C=O) and hydroxyl group (—OH). 16.2 IUPAC Nomenclature for Carboxylic Acids The naming of carboxylic acids is fairly simple. You simply find the longest carbon chain which includes the carboxylic group. Use that as the stem for the name, cross off the -e on the ending of the alkane name and replace it with -oic acid. I think you can see how that works, if you look at this example (which is also shown in Example 1-a in your workbook). It gives you, in this case propanoic acid (with a three-carbon-atom chain), the (from propan + oic acid) name propanoic acid. As with aldehydes, it is not necessary to indicate where the acid functional group is because it has to be at the end of the molecule, on the #1 carbon. -
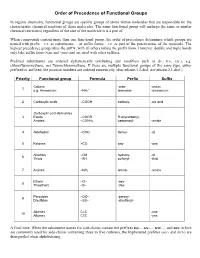
Priority of Functional Groups
Order of Precedence of Functional Groups In organic chemistry, functional groups are specific groups of atoms within molecules that are responsible for the characteristic chemical reactions of those molecules. The same functional group will undergo the same or similar chemical reaction(s) regardless of the size of the molecule it is a part of. When compounds contain more than one functional group, the order of precedence determines which groups are named with prefix – i.e. as substituents –, or suffix forms – i.e. as part of the parent name of the molecule. The highest precedence group takes the suffix, with all others taking the prefix form. However, double and triple bonds only take suffix form (-ene and -yne) and are used with other suffixes. Prefixed substituents are ordered alphabetically (excluding any modifiers such as di-, tri-, etc.), e.g. chlorofluoromethane, not fluorochloromethane. If there are multiple functional groups of the same type, either prefixed or suffixed, the position numbers are ordered numerically (thus ethane-1,2-diol, not ethane-2,1-diol.) Priority Functional group Formula Prefix Suffix Cations -onio- -onium 1 + e.g. Ammonium –NH4 ammonio- -ammonium 2 Carboxylic acids –COOH carboxy- -oic acid Carboxylic acid derivatives 3 Esters –COOR R-oxycarbonyl- Amides –CONH2 carbamoyl- -amide 4 Aldehydes –CHO formyl- -al 5 Ketones >CO oxo- -one Alcohols –OH hydroxy- -ol 6 Thiols –SH sulfanyl- -thiol 7 Amines –NH2 amino- -amine Ethers –O– -oxy- 8 Thioethers –S– -thio- Peroxides –OO– -peroxy- 9 Disulfides –SS– -disulfanyl- Alkenes C=C -ene 10 Alkynes C≡C -yne A final note: When the substituent names for side-chains contain the prefixes iso..., sec-..., tert-..., and neo (which are commonly used for side-chains containing three to five carbons), the hyphenated prefixes (sec- and tert-) are disregarded when alphabetizing. -

1 Chapter 3: Organic Compounds: Alkanes and Cycloalkanes
Chapter 3: Organic Compounds: Alkanes and Cycloalkanes >11 million organic compounds which are classified into families according to structure and reactivity Functional Group (FG): group of atoms which are part of a large molecule that have characteristic chemical behavior. FG’s behave similarly in every molecule they are part of. The chemistry of the organic molecule is defined by the function groups it contains 1 C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic C N C C C X X= F, Cl, Br, I amines Alkenes Alkyl Halide H C C C O C C O Alkynes alcohols ethers acidic H H H C S C C C C S C C H sulfides C C thiols (disulfides) H H Arenes Carbonyl-oxygen double bonds (carbonyls) Carbon-nitrogen multiple bonds acidic basic O O O N H C H C O C Cl imine (Schiff base) aldehyde carboxylic acid acid chloride O O O O C C N C C C C O O C C nitrile (cyano group) ketones ester anhydrides O C N amide opsin Lys-NH2 + Lys- opsin H O H N rhodopsin H 2 Alkanes and Alkane Isomers Alkanes: organic compounds with only C-C and C-H single (s) bonds. general formula for alkanes: CnH(2n+2) Saturated hydrocarbons Hydrocarbons: contains only carbon and hydrogen Saturated" contains only single bonds Isomers: compounds with the same chemical formula, but different arrangement of atoms Constitutional isomer: have different connectivities (not limited to alkanes) C H O C4H10 C5H12 2 6 O OH butanol diethyl ether straight-chain or normal hydrocarbons branched hydrocarbons n-butane n-pentane Systematic Nomenclature (IUPAC System) Prefix-Parent-Suffix -

Ch.04Carbon and the Molecular Diversity of Life
EXPERIMENT “Atmosphere” CH4 Water vapor Electrode Molecular Structural Ball-and-Stick Space-Filling Name Formula Formula Model Model NH 3 H 2 (a) Methane Condenser Cooled water (b) Ethane containing Cold organic water molecules (c) Ethene (ethylene) H2O “sea” Sample for chemical analysis 1 2 Hydrogen Oxygen Nitrogen Carbon (valence = 1) (valence = 2) (valence = 3) (valence = 4) H O N C Urea 3 4 Fat droplets (stained red) Ethane Propane 1-Butene 2-Butene (a) Length (c) Double bonds Butane 2-Methylpropane (commonly called isobutane) Cyclohexane Benzene (b) Branching (d) Rings 100 µm (a) Mammalian adipose cells (b) A fat molecule 5 6 Effective Ineffective Drug Condition Pentane 2-methyl butane Enantiomer Enantiomer (a) Structural isomers Ibuprofen Pain; inflammation cis isomer: The two Xs are trans isomer: The two Xs are S-Ibuprofen R-Ibuprofen on the same side. on opposite sides. (b) Geometric isomers Albuterol Asthma R-Albuterol S-Albuterol L isomer D isomer (c) Enantiomers 7 8 Estradiol Testosterone L-dopa D-dopa 9 10 CHEMICAL Hydroxyl Carbonyl Carboxyl CHEMICAL Amino Sulfhydryl Phosphate Methyl GROUP GROUP (may be STRUCTURE STRUCTURE written HS—) (may be written HO—) The amino group The sulfhydryl group In a phosphate group, a A methyl group consists of a In a hydroxyl group (—OH), a The carbonyl group ( CO) When an oxygen atom is (—NH2) consists of a consists of a sulfur atom phosphorus atom is bonded to carbon bonded to three hydrogen atom is bonded to an consists of a carbon atom double-bonded to a carbon nitrogen atom bonded bonded to an atom of four oxygen atoms; one oxygen hydrogen atoms. -

Chapter 3 Alcohols, Phenols, and Ethers
Chapter 3 Alcohols, Phenols, and Ethers Chapter 3 Alcohols, Phenols, and Ethers Chapter Objectives: • Learn to recognize the alcohol, phenol, and ether functional groups. • Learn the IUPAC system for naming alcohols, phenols, and ethers. • Learn the important physical properties of the alcohols, phenols, and ethers. • Learn the major chemical reaction of alcohols, and learn how to predict the products of dehydration and oxidation reactions. • Learn to recognize the thiol functional group. Mr. Kevin A. Boudreaux Angelo State University CHEM 2353 Fundamentals of Organic Chemistry Organic and Biochemistry for Today (Seager & Slabaugh) www.angelo.edu/faculty/kboudrea Introduction • In this chapter, we will start looking at organic molecules that incorporate C—O bonds. • Oxygen is in Group 6A of the periodic table, and in most of its compounds, contains two single bonds and two lone pairs (or one double bond and two lone pairs), and is sp3-hybridized with a bent molecular shape: O O •The alcohol, phenol, and ether functional groups are found in a number of important naturally occurring molecules: CH3CH2OH Ethanol OH CH3CH2OCH2CH3 Diethyl ether HO Menthol Cholesterol 2 1 Chapter 3 Alcohols, Phenols, and Ethers Alcohols 3 The Hydroxy (—OH) Functional Group •The hydroxyl group (—OH) is found in the alcohol and phenol functional groups. (Note: that’s not the same as hydroxide, OH-, which is ionic.) –in alcohols, a hydroxyl group is connected to a carbon atom. –in phenols, —OH is connected to a benzene ring. (The “parent” molecule of this class is also named phenol: PhOH or C6H5OH.) • When two carbon groups are connected by single bonds to an oxygen, this is classified as the ether functional group. -

Functional Group Interconversions 119
FUNCTIONAL GROUP INTERCONVERSIONS 119 Functional Group Interconversions C&S Chapter 3 #1; 2; 4a,b, e; 5a, b, d; 6a,b,c,d; 8 1 sulfonates 2 halides 3 nitriles 4 azides 5 amines 6 esters and lactones 7 amides and lactams Sulfonate Esters - reaction of an alcohols (1° or 2°) with a sulfonyl chloride O R'= R'SO2Cl CH3 mesylate R OH R O S R' triflate O CF3 sulfonate ester CH3 tosylate - sulfonate esters are very good leaving groups. Elimination is often a competing side reaction Halides - halides are good leaving groups with the order of reactivity in SN2 reactions being I>Br>Cl. Halides are less reactive than sulfonate esters, however elimination as a competing side reaction is also reduced. - sulfonate esters can be converted to halides with the sodium halide in acetone at reflux. Chlorides are also converted to either bromides or iodides in the same fashion (Finkelstein Reaction). O X- R O S R' R X X= Cl, Br, I O NaI, acetone reflux R Cl R I - conversion of hydroxyl groups to halides: Organic Reactions 1983, 29, 1 R OH R X - R-OH to R-Cl - SOCl2 - Ph3P, CCl4 - Ph3P, Cl2 - Ph3P, Cl3CCOCCl3 FUNCTIONAL GROUP INTERCONVERSIONS 120 - R-OH to R-Br - PBr3, pyridine - Ph3P, CBr4 - Ph3P, Br2 - R-OH to R-I - Ph3P, DEAD, MeI Nitriles - displacement of halides or sulfonates with cyanide anion KCN, 18-C-6 DMSO R X R C N - dehydration of amides O R C N R NH2 - POCl3, pyridine - TsCl, pyridine - P2O5 - SOCl2 - Reaction of esters and lactones with dimethylaluminium amide TL 1979, 4907 Me H3C Me2AlNH2 OH JOC 1987, 52, 1309 O NC Ar O Ar - Dehydration of oximes OH N H2NOH•HCl P2O5 R CHO R C N R H - Oxidation of hydrazones O O N C Tetrahedron Lett. -

3: Haloalkanes, Alcohols, Ethers, and Amines
(2/94)(2-4,9/95)(7/97)(11,12/98)(1,9,11/99) Neuman Chapter 3 3: Haloalkanes, Alcohols, Ethers, and Amines Halogen, OH, and NH2 Functional Groups Haloalkanes (RX) Alcohols (ROH) Ethers (ROR) Amines (RNH2, R2NH, R3N) Amines are Organic Bases Preview This chapter describes several classes of organic compounds with functional groups containing N, O, or halogen atoms (X) on their carbon skeletons. As a result, they have C-N, C-O, or C-X bonds (X = F, Cl, Br, or I) in addition to C-C and C-H bonds. These haloalkanes (RX), alcohols (ROH), ethers (ROR), and amines (RNH2, R2NH, R3N) have different properties than alkanes and cycloalkanes because the X, O, and N atoms have valence shell unshared electron pairs and their bonds to C are polar. We will also see that amines are organic bases because they react with both weak and strong acids. In order to discuss these acid/base reactions of amines, we review concepts of acidity and basicity in this chapter. 3.1 Halogen, OH, and NH2 Functional Groups Alkanes and cycloalkanes contain only C and H atoms, but most organic molecules also have other atoms such as N, O, and halogens (F, Cl, Br, and I). We will begin our study of these molecules by examining those containing fluorine (F), chlorine (Cl), bromine (Br) or iodine (I) (designated as X), as well as those with OH or NH2 groups. We write the general formulas of these compounds as R- X, R-OH, and R-NH2. Haloalkanes, Alcohols, and Amines (3.1A) The general formulas R-X, R-OH, and R-NH2 suggest two different ways to view these classes of compounds. -

Chapter 1 Organic Compounds: Alkanes
Chapter 1 Alkanes Chapter 1 Organic Compounds: Alkanes Chapter Objectives: • Learn the differences between organic and inorganic compounds. • Learn how to identify isomers of organic compounds. • Learn how to write condensed, expanded, and line structures for organic compounds. • Learn how to recognize the alkane functional group in organic compounds. • Learn the IUPAC system for naming alkanes and cycloalkanes. • Learn the important physical and chemical properties of the alkanes. Mr. Kevin A. Boudreaux Angelo State University CHEM 2353 Fundamentals of Organic Chemistry Organic and Biochemistry for Today (Seager & Slabaugh) www.angelo.edu/faculty/kboudrea Organic chemistry nowadays almost drives me mad. To me it appears like a primeval tropical forest full of the most remarkable things, a dreadful endless jungle into which one does not dare enter, for there seems to be no way out. Friedrich Wöhler 2 1 Chapter 1 Alkanes 3 What Do We Mean By “Organic”? • In everyday usage, the word organic can be found in several different contexts: – chemicals extracted from plants and animals were originally called “organic” because they came from living organisms. – organic fertilizers are obtained from living organisms. – organic foods are foods grown without the use of pesticides or synthetic fertilizers. • In chemistry, the words “organic” and “organic chemistry” are defined a little more precisely: 4 2 Chapter 1 Alkanes What is Organic Chemistry? • Organic chemistry is concerned with the study of the structure and properties of compounds containing carbon. – All organic compounds contain carbon atoms. – Inorganic compounds contain no carbons. Most inorganic compounds are ionic compounds. • Some carbon compounds are not considered to be organic (mostly for historical reasons), such as CO, CO2, diamond, graphite, and salts of carbon- 2- - containing polyatomic ions (e.g., CO3 , CN ). -

Structural Organic Chemistry the Shapes of Molecules Functional Groups Double (Four-Electron) Bond, and Three Lines a Triple (Six-Electron) Bond
STRUCTURAL ORGANIC CHEMISTRY. THE SHAPES OF MOLECULES. FUNCTIONAL GROUPS In this chapter we first briefly review the most important types of covalent bonds encountered in organic substances and the ways in which these bonds are represented in structural formulas. Next we consider the sizes and shapes of organic molecules and how structural formulas written in two dimensions can be translated into three-dimensional models that show the relative posi- tions of the atoms in space. We also discuss models that reflect the relative sizes of the atoms and the way in which the atoms may interfere with each other when in close quarters (steric hindrance). Then we go on to further im- portant aspects of structure-the functional group concept and position isomerism. Our aim is to have you become more familiar with the various kinds of organic compounds and begin to see how the practicing organic chemist visu- alizes molecules and correlates the diverse kinds of structures that he has to deal with in his work. 2-1 STRUCTURAL FORMULAS The building block of structural organic chemistry is the tetravalent carbon atom. With few exceptions, carbon compounds can be formulated with four 2-1 Structural Fbrmulas 31 covalent bonds to each carbon, regardless of whether the combination is with carbon or some other element. The two-electron bond, which is illustrated by the carbon-hydrogen bonds in methane or ethane and the carbon-carbon bond in ethane, is called a single bond. In these and many related substances, each carbon is attached to four other atoms: H:C:H. -

Methyl Groups Make a Late Entrance
News & views low because most people have the same low Monica Grant is in the Department of Acad. Sci. USA 113, 14294–14299 (2016). level of educational attainment. As access to Sociology, University of Wisconsin-Madison, 3. Lutz, W., Cuaresma, J. C. & Sanderson, W. Science 319, 1047–1048 (2008). schooling expands, inequality as measured Madison, Wisconsin 53706, USA. 4. Friedman, J. et al. Nature 580, 636–639 (2020). by the AID rises because the population now e-mail: [email protected] 5. Goujon, A. et al. J. Demogr. Econ. 82, 315–363 (2016). contains many people who have no education 6. Barro, R. J. & Lee, J. W. J. Dev. Econ. 104, 184–198 (2013). and many who have several years of schooling. 7. Jones, G. W. & Ramchand, D. S. J. Int. Dev. 28, 953–973 1. Marmot, M., Friel, S., Bell, R., Houweling, T. A. J. & (2016). As school enrolment becomes universal and Taylor, S. Lancet 372, 1661–1669 (2008). members of the population begin to achieve 2. Abel, G. J., Barakat, B., Samir, K. C. & Lutz, W. Proc. Natl This article was published online on 15 April 2020. similarly high levels of education, the AID declines again. From this metric, Friedman and Organic chemistry colleagues conclude that global educational inequality peaked in 2017 and is projected to decline until 2030. Methyl groups Even though this project involves an impressive volume of data, it is still limited by problems of data scarcity. Whereas some make a late entrance high-income countries, such as France and Germany, contribute more than 55 data Emily B.