3: Haloalkanes, Alcohols, Ethers, and Amines
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Relative Rates of Thiol–Thioester Exchange and Hydrolysis for Alkyl and Aryl Thioalkanoates in Water
Orig Life Evol Biosph (2011) 41:399–412 DOI 10.1007/s11084-011-9243-4 PREBIOTIC CHEMISTRY The Relative Rates of Thiol–Thioester Exchange and Hydrolysis for Alkyl and Aryl Thioalkanoates in Water Paul J. Bracher & Phillip W. Snyder & Brooks R. Bohall & George M. Whitesides Received: 14 April 2011 /Accepted: 16 June 2011 / Published online: 5 July 2011 # Springer Science+Business Media B.V. 2011 Abstract This article reports rate constants for thiol–thioester exchange (kex), and for acid- mediated (ka), base-mediated (kb), and pH-independent (kw) hydrolysis of S-methyl thioacetate and S-phenyl 5-dimethylamino-5-oxo-thiopentanoate—model alkyl and aryl thioalkanoates, respectively—in water. Reactions such as thiol–thioester exchange or aminolysis could have generated molecular complexity on early Earth, but for thioesters to have played important roles in the origin of life, constructive reactions would have needed to compete effectively with hydrolysis under prebiotic conditions. Knowledge of the kinetics of competition between exchange and hydrolysis is also useful in the optimization of systems where exchange is used in applications such as self-assembly or reversible binding. For the alkyl thioester S-methyl thioacetate, which has been synthesized in −5 −1 −1 −1 −1 −1 simulated prebiotic hydrothermal vents, ka = 1.5×10 M s , kb = 1.6×10 M s , and −8 −1 kw = 3.6×10 s . At pH 7 and 23°C, the half-life for hydrolysis is 155 days. The second- order rate constant for thiol–thioester exchange between S-methyl thioacetate and 2- −1 −1 sulfonatoethanethiolate is kex = 1.7 M s . -

Chapter 7, Haloalkanes, Properties and Substitution Reactions of Haloalkanes Table of Contents 1
Chapter 7, Haloalkanes, Properties and Substitution Reactions of Haloalkanes Table of Contents 1. Alkyl Halides (Haloalkane) 2. Nucleophilic Substitution Reactions (SNX, X=1 or 2) 3. Nucleophiles (Acid-Base Chemistry, pka) 4. Leaving Groups (Acid-Base Chemistry, pka) 5. Kinetics of a Nucleophilic Substitution Reaction: An SN2 Reaction 6. A Mechanism for the SN2 Reaction 7. The Stereochemistry of SN2 Reactions 8. A Mechanism for the SN1 Reaction 9. Carbocations, SN1, E1 10. Stereochemistry of SN1 Reactions 11. Factors Affecting the Rates of SN1 and SN2 Reactions 12. --Eliminations, E1 and E2 13. E2 and E1 mechanisms and product predictions In this chapter we will consider: What groups can be replaced (i.e., substituted) or eliminated The various mechanisms by which such processes occur The conditions that can promote such reactions Alkyl Halides (Haloalkane) An alkyl halide has a halogen atom bonded to an sp3-hybridized (tetrahedral) carbon atom The carbon–chlorine and carbon– bromine bonds are polarized because the halogen is more electronegative than carbon The carbon-iodine bond do not have a per- manent dipole, the bond is easily polarizable Iodine is a good leaving group due to its polarizability, i.e. its ability to stabilize a charge due to its large atomic size Generally, a carbon-halogen bond is polar with a partial positive () charge on the carbon and partial negative () charge on the halogen C X X = Cl, Br, I Different Types of Organic Halides Alkyl halides (haloalkanes) sp3-hybridized Attached to Attached to Attached -

Classifying Halogenoalkanes Reactions Of
10 Halogenoalkanes H Naming Halogenoalkanes H H H H C H H H H Based on original alkane, with a prefix indicating halogen atom: H C C C H Fluoro for F; Chloro for Cl; Bromo for Br; Iodo for I. H C C C C H Br H H H Cl H H Substituents are listed alphabetically 1-bromopropane 2-chloro-2-methylbutane Classifying Halogenoalkanes Halogenoalkanes can be classified as primary, secondary or tertiary depending on the number of carbon atoms attached to the C-X functional group. H H H H H H H H C H H H H H C C C H H C C C H H C C C C H Br H H H Br H H Cl H H Primary halogenoalkane Secondary halogenoalkane Tertiary halogenoalkane One carbon attached to the Two carbons attached to the Three carbons attached to the carbon atom adjoining the carbon atom adjoining the carbon atom adjoining the halogen halogen halogen Reactions of Halogenoalkanes Halogenoalkanes undergo either substitution or elimination reactions Nucleophilic substitution reactions Substitution: swapping a halogen atom for another atom or groups of atoms - - Nucleophile: electron pair donator e.g. :OH , :NH3, CN :Nu represents any nucleophile – they The Mechanism: We draw (or outline) mechanisms to always have a lone pair and act as show in detail how a reaction proceeds electron pair donators Nu:- The nucleophiles The carbon has a small attack the positive H H H H positive charge because carbon atom of the electronegativity H C C X H C C + X- δ+ δ- Nu difference between the carbon and the halogen H H H H We use curly arrows in mechanisms (with A curly arrow will always two line heads) to show the movement of start from a of electrons or two electrons lone pair the centre of a bond The rate of these substitution reactions depends on the strength Bond enthalpy / of the C-X bond kJmol-1 The weaker the bond, the easier it is to break and the faster the reaction. -
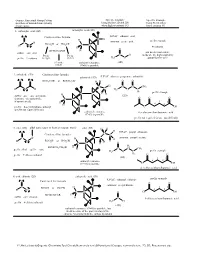
Generic Functional Group Pattern in Ordero of Nomenclature Priority in Our Course. Specific Example Using Suffix (2D and 3D)
Generic Functional Group Pattern Specific Example Specific Example in ordero of nomenclature priority Using Suffix (2D and 3D) Using Prefix when in our course. when highest priority FG lower priority FG 1. carboxylic acid (2D) carboxylic acid (3D) O Condensed line formula IUPAC: ethanoic acid O H H prefix example C H common: acetic acid RCO2H or HO2CR C C R O #-carboxy H O RCH(CO H)R' H suffix: -oic acid 2 O not used in our course FG on FG on C H (acids are the highest priority prefix: #-carboxy the right the left H3C O group that we use) FG in the carbonyl resonance (2D) middle (C=O) is possible 2. anhydride (2D) Condensed line formula anhydride (3D) IUPAC: ethanoic propanoic anhydride O O RCO2COR' or ROCO2CR' H H O O H C C C CH C 3 R O R H3C O C H H 2 prefix example H O C H suffix: -oic -oic anhydride (2D) (just one -oic anhydride, O O O C C O if symmetrical) 3 1 H 4 H 2 prefix: #-acyloxyalkanecarbonyl O O OH (prefix not required for us) carbonyl resonance 4-acyloxymethanebutanoic acid (C=O) is possible (prefix not required for us - too difficult) 3. ester (2D) alkyl name (goes in front as separate word) ester (3D) H H H IUPAC: propyl ethanoate O C Condensed line formula C O C common: propyl acetate C R' H H R O RCO2R' or R'O2CR H H C C H O H H2 RCH(CO2CH3)R' prefix: alkyl suffix: -oate H C C CH3 O H C O C prefix example 3 H prefix: #-alkoxycarbonyl 2 O O (2D) 3 1 carbonyl resonance 4 2 (C=O) is possible O OH 4-methoxycarbonylbutanoic acid 4. -

Nomenclature of Carboxylic Acids • Select the Longest Carbon Chain Containing the Carboxyl Group
Chapter 5 Carboxylic Acids and Esters Carboxylic Acids • Carboxylic acids are weak organic acids which Chapter 5 contain the carboxyl group (RCO2H): Carboxylic Acids and Esters O C O H O RCOOH RCO2H Chapter Objectives: O condensed ways of • Learn to recognize the carboxylic acid, ester, and related functional groups. RCOH writing the carboxyl • Learn the IUPAC system for naming carboxylic acids and esters. group a carboxylic acid C H • Learn the important physical properties of the carboxylic acids and esters. • Learn the major chemical reaction of carboxylic acids and esters, and learn how to O predict the products of ester synthesis and hydrolysis reactions. the carboxyl group • Learn some of the important properties of condensation polymers, especially the polyesters. Mr. Kevin A. Boudreaux • The tart flavor of sour-tasting foods is often caused Angelo State University CHEM 2353 Fundamentals of Organic Chemistry by the presence of carboxylic acids. Organic and Biochemistry for Today (Seager & Slabaugh) www.angelo.edu/faculty/kboudrea 2 Nomenclature of Carboxylic Acids • Select the longest carbon chain containing the carboxyl group. The -e ending of the parent alkane name is replaced by the suffix -oic acid. • The carboxyl carbon is always numbered “1” but the number is not included in the name. • Name the substituents attached to the chain in the Nomenclature of usual way. • Aromatic carboxylic acids (i.e., with a CO2H Carboxylic Acids directly connected to a benzene ring) are named after the parent compound, benzoic acid. O C OH 3 -

Introduction to Alkenes and Alkynes in an Alkane, All Covalent Bonds
Introduction to Alkenes and Alkynes In an alkane, all covalent bonds between carbon were σ (σ bonds are defined as bonds where the electron density is symmetric about the internuclear axis) In an alkene, however, only three σ bonds are formed from the alkene carbon -the carbon thus adopts an sp2 hybridization Ethene (common name ethylene) has a molecular formula of CH2CH2 Each carbon is sp2 hybridized with a σ bond to two hydrogens and the other carbon Hybridized orbital allows stronger bonds due to more overlap H H C C H H Structure of Ethylene In addition to the σ framework of ethylene, each carbon has an atomic p orbital not used in hybridization The two p orbitals (each with one electron) overlap to form a π bond (p bonds are not symmetric about the internuclear axis) π bonds are not as strong as σ bonds (in ethylene, the σ bond is ~90 Kcal/mol and the π bond is ~66 Kcal/mol) Thus while σ bonds are stable and very few reactions occur with C-C bonds, π bonds are much more reactive and many reactions occur with C=C π bonds Nomenclature of Alkenes August Wilhelm Hofmann’s attempt for systematic hydrocarbon nomenclature (1866) Attempted to use a systematic name by naming all possible structures with 4 carbons Quartane a alkane C4H10 Quartyl C4H9 Quartene e alkene C4H8 Quartenyl C4H7 Quartine i alkine → alkyne C4H6 Quartinyl C4H5 Quartone o C4H4 Quartonyl C4H3 Quartune u C4H2 Quartunyl C4H1 Wanted to use Quart from the Latin for 4 – this method was not embraced and BUT has remained Used English order of vowels, however, to name the groups -

A Perovskite-Based Paper Microfluidic Sensor for Haloalkane Assays
ORIGINAL RESEARCH published: 26 April 2021 doi: 10.3389/fchem.2021.682006 A Perovskite-Based Paper Microfluidic Sensor for Haloalkane Assays Lili Xie 1, Jie Zan 1, Zhijian Yang 1, Qinxia Wu 1, Xiaofeng Chen 1, Xiangyu Ou 1, Caihou Lin 2*, Qiushui Chen 1,3* and Huanghao Yang 1,3* 1 Ministry of Education (MOE) Key Laboratory for Analytical Science of Food Safety and Biology, Fujian Provincial Key Laboratory of Analysis and Detection Technology for Food Safety, College of Chemistry, Fuzhou University, Fuzhou, China, 2 Department of Neurosurgery, Fujian Medical University Union Hospital, Fuzhou, China, 3 Fujian Science and Technology Innovation Laboratory for Optoelectronic Information of China, Fuzhou, China Detection of haloalkanes is of great industrial and scientific importance because some Edited by: haloalkanes are found serious biological and atmospheric issues. The development Jinyi Lin, of a flexible, wearable sensing device for haloalkane assays is highly desired. Here, Nanjing Tech University, China we develop a paper-based microfluidic sensor to achieve low-cost, high-throughput, Reviewed by: Xiaobao Xu, and convenient detection of haloalkanes using perovskite nanocrystals as a nanoprobe Nanjing University of Science and through anion exchanging. We demonstrate that the CsPbX3 (X = Cl, Br, or I) Technology, China nanocrystals are selectively and sensitively in response to haloalkanes (CH2Cl2, CH2Br2), Xuewen Wang, Northwestern Polytechnical and their concentrations can be determined as a function of photoluminescence University, China spectral shifts of perovskite nanocrystals. In particular, an addition of nucleophilic trialkyl *Correspondence: phosphines (TOP) or a UV-photon-induced electron transfer from CsPbX3 nanocrystals is Caihou Lin [email protected] responsible for achieving fast sensing of haloalkanes. -

Linear Alkylbenzene Sulphonate (CAS No
Environmental Risk Assessment LAS Linear Alkylbenzene Sulphonate (CAS No. 68411-30-3) Revised ENVIRONMENTAL Aspect of the HERA Report = February 2013 = 1 1. Contents 2. Executive summary 3. Substance characterisation 3.1 CAS No. and grouping information 3.2 Chemical structure and composition 3.3 Manufacturing route and production/volume statistics 3.4 Consumption scenario in Europe 3.5 Use application summary 4. Environmental safety assessment 4.1 Environmental exposure assessment 4.1.1 Biotic and abiotic degradability 4.1.2 Removal 4.1.3 Monitoring studies 4.1.4 Exposure assessment: scenario description 4.1.5 Substance data used for the exposure calculation 4.1.6 PEC calculations 4.1.7 Bioconcentration 4.2 Environmental effects assessment 4.2.1 Ecotoxicity 4.2.1.1 Aquatic ecotoxicity 4.2.1.2 Terrestrial ecotoxicity 4.2.1.3 Sediment ecotoxicity 4.2.1.4 Ecotoxicity to sewage microorganisms 4.2.1.5 Reassurance on absence of estrogenic effects 4.2.2 PNEC calculations 4.2.2.1 Aquatic PNEC 4.2.2.2 Terrestrial PNEC 4.2.2.3 Sludge PNEC 4.2.2.4 Sediment PNEC 4.2.2.5 STP PNEC 4.3 Environment risk assessment 5. 5. References 6. Contributors to the report 6.1 Substance team 6.2 HERA environmental task force 6.3 HERA human health task force 6.4 Industry coalition for the OECD/ICCA SIDS assessment of LAS 2 2. Executive Summary Linear alkylbenzene sulphonate (LAS) is an anionic surfactant. It was introduced in 1964 as the readily biodegradable replacement for highly branched alkylbenzene sulphonates (ABS). -

Chapter 7 - Alkenes and Alkynes I
Andrew Rosen Chapter 7 - Alkenes and Alkynes I 7.1 - Introduction - The simplest member of the alkenes has the common name of ethylene while the simplest member of the alkyne family has the common name of acetylene 7.2 - The (E)-(Z) System for Designating Alkene Diastereomers - To determine E or Z, look at the two groups attached to one carbon atom of the double bond and decide which has higher priority. Then, repeat this at the other carbon atom. This system is not used for cycloalkenes - If the two groups of higher priority are on the same side of the double bond, the alkene is designated Z. If the two groups of higher priority are on opposite sides of the double bond, the alkene is designated E - Hydrogenation is a syn/cis addition 7.3 - Relative Stabilities of Alkenes - The trans isomer is generally more stable than the cis isomer due to electron repulsions - The addition of hydrogen to an alkene, hydrogenation, is exothermic (heat of hydrogenation) - The greater number of attached alkyl groups, the greater the stability of an alkene 7.5 - Synthesis of Alkenes via Elimination Reactions, 7.6 - Dehydrohalogenation of Alkyl Halides How to Favor E2: - Reaction conditions that favor elimination by E1 should be avoided due to the highly competitive SN 1 mechanism - To favor E2, a secondary or tertiary alkyl halide should be used - If there is only a possibility for a primary alkyl halide, use a bulky base - Use a higher concentration of a strong and nonpolarizable base, like an alkoxide - EtONa=EtOH favors the more substituted double bond -

Educational Research Applications Abebe M, Et Al
Educational Research Applications Abebe M, et al. Educ Res Appl 5: 175. Review Article DOI: 10.29011/2575-7032.100175 Teaching Students Synthesizing Molecules Mimicking an Existing Drug against Covid-19 Moges Abebe1*, Lashan Eloise Knowles1, Bisrat Hailemeskel2 1Department of Biological and Physical Sciences, Saint Augustine University, Raleigh, NC, USA 2Department of Clinical & Administrative Pharmacy Sciences, College of Pharmacy, Howard University, NW Washington, DC, USA *Corresponding author: Moges Abebe, Department of Biological and Physical Sciences, Saint Augustine University, Raleigh, NC 27610, NC, USA Citation: Abebe M, Knowles LE, Hailemeskel B (2020) Teaching Students Synthesizing Molecules Mimicking an Existing Drug against Covid-19. Educ Res Appl 5: 175. DOI: 10.29011/2575-7032.100175 Received Date: 26 May 2020; Accepted Date: 01 June, 2020; Published Date: 06 June, 2020 Abstract End of semester organic chemistry course projects are valuable learning assessment tools while giving students a creative opportunity and sparking interest for further research investigations. The purpose of this year’s project was to teach students how to synthesize a molecule that potentially mimics an existing drug that works against the COVID-19. The available drugs chosen for the project are those that are proposed to work either by prohibiting the easy entry of the virus into respiratory tissues or those who deprive the virus’s ability to reproduce once they enter the cell. An investigative search in historical literature and the current conditions of the virus enabled students to create a unique and innovative product that requires a cumulative learned knowledge. History has shown that when a new virus becomes pandemic it takes time for researchers to create a drug, test the results, and gets approved by the Food and Drug Administration (FDA) for public availability. -

Reactions of Alkenes and Alkynes
05 Reactions of Alkenes and Alkynes Polyethylene is the most widely used plastic, making up items such as packing foam, plastic bottles, and plastic utensils (top: © Jon Larson/iStockphoto; middle: GNL Media/Digital Vision/Getty Images, Inc.; bottom: © Lakhesis/iStockphoto). Inset: A model of ethylene. KEY QUESTIONS 5.1 What Are the Characteristic Reactions of Alkenes? 5.8 How Can Alkynes Be Reduced to Alkenes and 5.2 What Is a Reaction Mechanism? Alkanes? 5.3 What Are the Mechanisms of Electrophilic Additions HOW TO to Alkenes? 5.1 How to Draw Mechanisms 5.4 What Are Carbocation Rearrangements? 5.5 What Is Hydroboration–Oxidation of an Alkene? CHEMICAL CONNECTIONS 5.6 How Can an Alkene Be Reduced to an Alkane? 5A Catalytic Cracking and the Importance of Alkenes 5.7 How Can an Acetylide Anion Be Used to Create a New Carbon–Carbon Bond? IN THIS CHAPTER, we begin our systematic study of organic reactions and their mecha- nisms. Reaction mechanisms are step-by-step descriptions of how reactions proceed and are one of the most important unifying concepts in organic chemistry. We use the reactions of alkenes as the vehicle to introduce this concept. 129 130 CHAPTER 5 Reactions of Alkenes and Alkynes 5.1 What Are the Characteristic Reactions of Alkenes? The most characteristic reaction of alkenes is addition to the carbon–carbon double bond in such a way that the pi bond is broken and, in its place, sigma bonds are formed to two new atoms or groups of atoms. Several examples of reactions at the carbon–carbon double bond are shown in Table 5.1, along with the descriptive name(s) associated with each. -

In This Handout, All of Our Functional Groups Are Presented As Condensed Line Formulas, 2D and 3D Formulas and with Nomenclature Prefixes and Suffixes (If Present)
In this handout, all of our functional groups are presented as condensed line formulas, 2D and 3D formulas and with nomenclature prefixes and suffixes (if present). Organic names are built on a foundation of alkanes, alkenes and alkynes. Those examples are presented first and you need to know those rules. The strategies can be found in Chapter 4 of our textbook (alkanes: pages 93-98, cycloalkanes 102-104, alkenes: pages 104-110, alkynes: pages 112-113 and combinations of all of them 113-115). After introducing examples of alkanes, alkenes, alkynes and combinations of them, the functional groups are presented in order of priority. A few nomenclature examples are provided for each of the functional groups. Examples of the various functional groups are presented on pages 115-135 in the textbook. Two overview pages are on pages 136-137. Some functional groups have a suffix name when they are the highest priority functional group and a prefix name when they are not the highest priority group, and these are added to the skeletal names with identifying numbers and stereochemistry terms (E and Z for alkenes, R and S for chiral centers and cis and trans for rings). Several low priority functional groups only have a prefix name. A few additional special patterns are shown on pages 98-102. The only way to learn this topic is practice (over and over). The best practice approach is to actually write out the names (on an extra piece of paper or on a white board, and then do it again). The same functional groups are used throughout the entire course.