Ketones and Aldehydes
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Functional Group Composition of Secondary Organic Aerosol Formed from Ozonolysis of A-Pinene Under High VOC and Autoxidation
Article Cite This: ACS Earth Space Chem. 2018, 2, 1196−1210 http://pubs.acs.org/journal/aesccq Functional Group Composition of Secondary Organic Aerosol Formed from Ozonolysis of α‑Pinene Under High VOC and Autoxidation Conditions Megan S. Claflin,†,‡ Jordan E. Krechmer,†,‡,§ Weiwei Hu,†,‡,∥ Jose L. Jimenez,†,‡ and Paul J. Ziemann*,†,‡ †Cooperative Institute for Research in Environmental Sciences (CIRES), Boulder, Colorado 80309, United States ‡Department of Chemistry and Biochemistry, University of Colorado, Boulder, Colorado 80309, United States *S Supporting Information ABSTRACT: The formation of secondary organic aerosol (SOA) from α-pinene ozonolysis has been widely studied, with a recent focus on contributions from highly oxidized multifunctional compounds (HOMs) that have been observed in laboratory and field studies. Most of what is known about the chemical composition of SOA and HOMs, however, consists of molecular formulas and limited molecular structure identification based on mass spectrometric analysis. Here, we characterized the SOA formed from α-pinene ozonolysis using derivatization-spectrophotometric methods to quantify per- oxide, carbonyl, carboxyl, ester, and hydroxyl groups. Experiments were conducted over a range of α-pinene concentrations and relative humidities, including regimes in − which gas-phase HOMs were detected using NO3 chemical ionization mass spectrometry. Results for experiments conducted with high concentrations of α-pinene were also compared with predictions of a model that employed the Master Chemical Mechanism and included gas-particle and gas-wall partitioning. It appears that gas-phase monomer and dimer products formed • • • • through RO2 +RO2 ,RO2 +HO2,RO2 isomerization, and stabilized Criegee intermediate + carboxylic acid or water reactions contributed to SOA formation, but that in particles the aldehyde and ketone groups in these compounds were often converted to carboxyl and ester groups through Baeyer−Villiger reactions with hydroperoxides and peroxycarboxylic acids. -
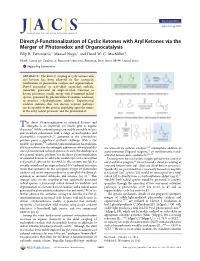
Direct Β‑Functionalization of Cyclic Ketones with Aryl Ketones Via the Merger of Photoredox and Organocatalysis Filip R
Communication pubs.acs.org/JACS Direct β‑Functionalization of Cyclic Ketones with Aryl Ketones via the Merger of Photoredox and Organocatalysis Filip R. Petronijevic,́† Manuel Nappi,† and David W. C. MacMillan* Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States *S Supporting Information ABSTRACT: The direct β-coupling of cyclic ketones with aryl ketones has been achieved via the synergistic combination of photoredox catalysis and organocatalysis. Diaryl oxymethyl or aryl−alkyl oxymethyl radicals, transiently generated via single-electron reduction of ketone precursors, readily merge with β-enaminyl radical species, generated by photon-induced enamine oxidation, to produce γ-hydroxyketone adducts. Experimental evidence indicates that two discrete reaction pathways can be operable in this process depending upon the nature of the ketyl radical precursor and the photocatalyst. he direct β-functionalization of saturated ketones and T aldehydes is an important yet elusive goal in organic chemistry.1 While carbonyl groups are readily amenable to ipso- and α-carbon substitution with a range of nucleophiles and electrophiles respectively,2,3 activation at the β-methylene position poses a significant synthetic challenge. With a few notable exceptions,1,4 carbonyl β-functionalization has tradition- ally been restricted to the conjugate addition of soft nucleophiles are accessed via carbene catalysis,9,10 nucleophilic addition of into α,β-unsaturated carbonyl systems. As such, the development acetal-protected Grignard reagents,11 or stoichiometric metal- 5 − of a general catalytic platform for the direct β-functionalization activated homoenolate equivalents.12 16 of saturated ketones or aldehydes would represent a conceptual Drawing from the mechanistic insights gained in the course of and practical advance for the field. -

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

Aldehydes and Ketones
12 Aldehydes and Ketones Ethanol from alcoholic beverages is first metabolized to acetaldehyde before being broken down further in the body. The reactivity of the carbonyl group of acetaldehyde allows it to bind to proteins in the body, the products of which lead to tissue damage and organ disease. Inset: A model of acetaldehyde. (Novastock/ Stock Connection/Glow Images) KEY QUESTIONS 12.1 What Are Aldehydes and Ketones? 12.8 What Is Keto–Enol Tautomerism? 12.2 How Are Aldehydes and Ketones Named? 12.9 How Are Aldehydes and Ketones Oxidized? 12.3 What Are the Physical Properties of Aldehydes 12.10 How Are Aldehydes and Ketones Reduced? and Ketones? 12.4 What Is the Most Common Reaction Theme of HOW TO Aldehydes and Ketones? 12.1 How to Predict the Product of a Grignard Reaction 12.5 What Are Grignard Reagents, and How Do They 12.2 How to Determine the Reactants Used to React with Aldehydes and Ketones? Synthesize a Hemiacetal or Acetal 12.6 What Are Hemiacetals and Acetals? 12.7 How Do Aldehydes and Ketones React with CHEMICAL CONNECTIONS Ammonia and Amines? 12A A Green Synthesis of Adipic Acid IN THIS AND several of the following chapters, we study the physical and chemical properties of compounds containing the carbonyl group, C O. Because this group is the functional group of aldehydes, ketones, and carboxylic acids and their derivatives, it is one of the most important functional groups in organic chemistry and in the chemistry of biological systems. The chemical properties of the carbonyl group are straightforward, and an understanding of its characteristic reaction themes leads very quickly to an understanding of a wide variety of organic reactions. -

Aldehydes and Ketones Are Simple Organic Compounds Containing a Carbonyl Group
Aldehydes and Ketones are simple organic compounds containing a carbonyl group. Carbonyl group contains carbon- oxygen double bond. These organic compounds are simple because the carbon atom presents in the carbonyl group lack reactive groups such as OH or Cl. By Dr. Sayed Hasan Mehdi Assistant Professor Department of Chemistry Shia P.G. College, Lucknow Dr. S.Hasan Mehdi 6/13/2020 This is to bring to kind notice that the matter of this e- content is for the B.Sc. IV semester students. It has been taken from the following sources. The students are advised to follow these books as well. •A TEXTBOOK OF ORGANIC CHEMISTRY by Arun Bahl & B.S. Bahl, S. Chand & Company Ltd. Publication. •Graduate Organic Chemistry by M. K. Jain and S.C. Sharma, Vishal Publishing Co. •Pradeep’s Organic Chemistry Vol II by R. N. Dhawan, Pradeep Publication, Jalandhar. Dr. S.Hasan Mehdi 6/13/2020 An aldehyde is one of the classes of carbonyl group- containing alkyl group on one end and hydrogen on the other end. The R and Ar denote alkyl or aryl member respectively. In the condensed form, the aldehyde is written as –CHO. Dr. S.Hasan Mehdi 6/13/2020 Dr. S.Hasan Mehdi 6/13/2020 1. From Alcohols: a. By oxidation of Alcohols: Aldehydes and ketones can be prepared by the controlled oxidation of primary and secondary alcohols using an acidified solution of potassium dichromate or permanganate. Primary alcohol produces aldehydesRef. Last slide. O K Cr O RCH2OH + [O] 2 2 7 + R C H H 10 Alcohol Aldehyde O CH3CH2OH+ [O] K2Cr2O7 + CH3 C H H Ethyl Alcohol Acetaldehyde The aldehydes formed in the above reaction are very easily oxidised to carboxylc acids if allowed to remain in the reaction mixture. -

Chemistry 0310 - Organic Chemistry 1 Chapter 6
Dr. Peter Wipf Chemistry 0310 - Organic Chemistry 1 Chapter 6. SN2 Reactions Nucleophilic substitution reactions occur when nucleophiles (electron-rich species) displace leaving groups on electrophiles (electron-poor species). The net result is the substitution of one group for another bonded to a carbon atom. Nucleophilicity is increased by a negative charge and an increase in the polarizability. The greater the size and the lower the electronegativity of an atom, the greater its polarizability. Leaving groups are stable species that can be detached with their bonding electrons from a molecule during reaction. Most good leaving groups are the conjugate bases of strong acids. Sulfonates (mesylates, tosylates, triflates, etc.) are popular leaving groups since they can be readily obtained from alcohols. Solvents also influence nucleophilicity. SN2 reactions are second-order reactions whose rates depend o the concentration of both the alkyl halide and the nucleophile. The transition state of the one-step SN2 process involves a trigonal bipyramidal carbon and results in an overall inversion of configuration. The order of reactivity of alkyl halides and alkyl sulfonates in SN2 reactions is CH3>1°>2°>3°. The activation energy, DG‡, determines the rate of a reaction: k = koexp(-DG‡/RT). The overall change in free energy, DG, determines the position of the thermodynamic equilibrium. A positive sign of DG is characteristic for an endergonic reaction, a negative DG is found for an exergonic process. The rate of a reaction can be measured and provides information about its mechanism. The reaction order is the sum of the exponents in the rate equation. Relative Leaving Group Abilities: Leaving Group Common Name krel AcO acetate 1 x 10-10 Cl chloride 0.0001 Br bromide 0.001 I iodide 0.01 O mesylate 1.00 H3C S O O O tosylate 0.70 H3C S O O O brosylate 2.62 Br S O O O nosylate 13 O2N S O O O fluorosulfate 29,000 F S O O O triflate 56,000 CF3 S O O O nonaflate 120,000 C4F9 S O O. -

The Reactions of Alkenes
The Reactions of Alkenes The Stereochemistry of Addition Reactions 1 Diverse Reactions of Alkenes Alkenes react with many electrophiles to give useful products by addition (often through special reagents) 2 Preparation of Alkenes: A Preview of Elimination Reactions • Alkenes are commonly made by – elimination of HX from alkyl halide (dehydrohalogenation) • Uses heat and KOH – elimination of H-OH from an alcohol (dehydration) • requires strong acids (sulfuric acid, 50 ºC) 3 A Regioselective Reaction A reaction in which one structural isomer is favored over another, leading to its predominance in the mixture of products. 4 A Stereoselective Reaction A reaction in which one stereoisomer in a mixture is produced more rapidly than another, resulting in predominance of the favored stereoisomer in the mixture of products. 5 A Stereospecific Reaction A reaction in which a particular stereoisomeric form of reactant gives one specific stereoisomer of product, while a different stereoisomeric form of reactant leads to a different single pure streoisomer of product. Stereospecific reaction is also stereoselective; however, stereoselective reaction is not stereospecific. 6 An Electrophilic Addition Reaction where HX = HF, HCl, HBr, and HI Reactivity of HF << HCl < HBr < HI since HF is less acidic and HI is most acidic. The rate of addition of HI is too fast to measure. 7 The Mechanism of the Reaction 8 Relative Stabilities of Carbocations 9 Hyperconjugation Stabilizes a Carbocation 10 The Difference in Carbocation Stability Determines the Products -

Stereochemistry, Alkyl Halide Substitution (SN1 & SN2)
Chem 261 Assignment & Lecture Outline 3: Stereochemistry, Alkyl Halide Substitution (SN1 & SN2) Read Organic Chemistry, Solomons, Fryle & Snyder 12th Edition (Electronic) • Functional Group List – Learn to recognize – Please see Green Handout – also p 76 of text • Periodic Table – Inside Front Cover - know 1st 10 elements (up through Neon) • Relative Strength of Acids and Bases – Inside Front cover (reference only) • Chapter 5 – Stereochemistry • Chapter 6 –Ionic Reactions: Nucleophilic Substitution of Alkyl Halides Problems: Do Not turn in, answers available in "Study Guide Student Solutions Manual " Solomons, Fryle, Snyder • Chapter 5: 5.1 to 5.15; 5.18 to 5.21; 5.26; 5.28; 5.33a-d; 5.46 • Chapter 6: 6.1 to 6.5; 6.7 to 6.10; 6.13; 6.20; 6.26; 6.27 Lecture Outline # 3 I. Comparison of 2 Structures: Same Molecular Formula ? -> If Yes, Possibly Isomers or Identical Same Arrangement (Sequence) of Groups ? If No -> Structural Isomers If Yes -> Superposable? If Yes -> Identical Structures If No -> Stereoisomers Non-Superposable Mirror Images ? If NO -> Diastereomers If Yes -> Enantiomers II. Chirality and Stereoisomers A. The Concept of Chirality 1. Identification of chiral objects a) achiral = not chiral b) planes of symmetry within a molecule 2. Types of stereoisomers – enantiomers and diastereomers B. Location of stereogenic (chiral) centres – 4 different groups on tetrahedral atom 1. Enantiomers & diastereomers 2. Meso compounds - chiral centers with plane of symmetry within molecule 3. Molecules with more than one chiral centre 4. Recognition of chiral centers in complex molecules - cholesterol - 8 chiral centres Drawing the enantiomer of cholesterol and its potential 255 stereoisomers 5. -

Chapter 14 – Aldehydes and Ketones
Chapter 14 – Aldehydes and Ketones 14.1 Structures and Physical Properties of Aldehydes and Ketones Ketones and aldehydes are related in that they each possess a C=O (carbonyl) group. They differ in that the carbonyl carbon in ketones is bound to two carbon atoms (RCOR’), while that in aldehydes is bound to at least one hydrogen (H2CO and RCHO). Thus aldehydes always place the carbonyl group on a terminal (end) carbon, while the carbonyl group in ketones is always internal. Some common examples include (common name in parentheses): O O H HH methanal (formaldehyde) trans-3-phenyl-2-propenal (cinnamaldehyde) preservative oil of cinnamon O O propanone (acetone) 3-methylcyclopentadecanone (muscone) nail polish remover a component of one type of musk oil Simple aldehydes (e.g. formaldehyde) typically have an unpleasant, irritating odor. Aldehydes adjacent to a string of double bonds (e.g. 3-phenyl-2-propenal) frequently have pleasant odors. Other examples include the primary flavoring agents in oil of bitter almond (Ph- CHO) and vanilla (C6H3(OH)(OCH3)(CHO)). As your book says, simple ketones have distinctive odors (similar to acetone) that are typically not unpleasant in low doses. Like aldehydes, placing a collection of double bonds adjacent to a ketone carbonyl generally makes the substance more fragrant. The primary flavoring agent in oil of caraway is just a such a ketone. 2 O oil of carraway Because the C=O group is polar, small aldehydes and ketones enjoy significant water solubility. They are also quite soluble in typical organic solvents. 14.2 Naming Aldehydes and Ketones Aldehydes The IUPAC names for aldehydes are obtained by using rules similar to those we’ve seen for other functional groups (e.g. -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -

Carbonyl Compounds
CARBONYL COMPOUNDS PART-4, PPT-4, SEM-3 Dr. Kalyan Kumar Mandal Associate Professor St. Paul’s C. M. College Kolkata CONTENTS: CARBONYL COMPOUNDS PART-4 • Formation of Acetal/Ketal • Formation of Thioacetal Reaction of Carbonyl Compounds with Alcohols • Carbonyl compounds react with alcohols. The product of this reaction is known as a hemiacetal, because it is halfway to an acetal. This reaction is analogous to hydrate formation from aldehydes and ketones. The mechanism follows in the footsteps of hydrate formation: ROH is used instead of HOH (water). This Lecture is prepared by Dr. K. K. Mandal, SPCMC, Kolkata Formation of Cyclic Hemiacetal • Hemiacetal formation is reversible, and they are stabilized by the same special structural features as those of hydrates. However, hemiacetals can also gain stability by being cyclic. • Cyclic hemiacetal is formed when the carbonyl group and the attacking hydroxyl group are part of the same molecule. The reaction is an intramolecular (within the same molecule) addition, as opposed to the intermolecular (between two molecules) ones. This is an example of ring-chain tautomerism. This Lecture is prepared by Dr. K. K. Mandal, SPCMC, Kolkata Formation of Cyclic Hemiacetal • Although the cyclic hemiacetal is more stable, it is still in equilibrium with some of the open-chain hydroxyaldehyde form. Its stability, and how easily it forms, depend on the size of the ring. • Five- and six-membered rings involve less strain (their bonds are free to adopt 109° or 120° angles) in comparison to the three- membered rings, and therefore five or six-membered hemiacetals are very common. -

Leaving Group of Substrate Important
1 Recap... • Last two lectures we looked at substitution reactions • We found that there we two (extreme) mechanisms • SN1 H H H H H PhS R H R Br R SPh Br • And SN2 H H H H PhS + Br R Br PhS R • We looked at many of the factors that influenced which ..mechanism was operating... • Now we will turn our attention to elimination reactions 2 Elimination reactions NaOH NaOH or low Br H2O OH concentration • You have seen SN1 substitution • Reaction not sped up by altering nucleophile - it is not in RDS • In fact if we increase the amount / concentration of nucleophile we get the following... high O H + + + Br Br OH concentration H H • We observe an elimination reaction • Overall HBr is lost from the molecule • Isn't chemistry fun - these are the same conditions as substitution! • Next two lectures will look at the mechanisms for elimination • And how we control the nature of the reaction we observe... 3 Elimination, bimolecular E2 O H Br HO Br H H • E2 - Elimination bimolecular or 2nd order reaction • 2 molecules in the RDS or transition state H H H H H C H C H H C Br Br H O H C H O H C C H H H H C H C H H H • Two molecules in the rate determining step • So both the base and the substrate control the reaction • Both carbon skeleton & leaving group of substrate important In substitutions nucleophile acts as a nucleophile & attacks carbon In eliminations nucleophile acts as a base & attacks proton 4 Elimination, bimolecular E2: MO • Little confusing as need bonding & anti-bonding in same diagram! • Orbitals need to be parallel for maximum overlap