Genomic Imbalances Pinpoint Potential Oncogenes and Tumor Suppressors in Wilms Tumors A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

The Landscape of Human Mutually Exclusive Splicing
bioRxiv preprint doi: https://doi.org/10.1101/133215; this version posted May 2, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. The landscape of human mutually exclusive splicing Klas Hatje1,2,#,*, Ramon O. Vidal2,*, Raza-Ur Rahman2, Dominic Simm1,3, Björn Hammesfahr1,$, Orr Shomroni2, Stefan Bonn2§ & Martin Kollmar1§ 1 Group of Systems Biology of Motor Proteins, Department of NMR-based Structural Biology, Max-Planck-Institute for Biophysical Chemistry, Göttingen, Germany 2 Group of Computational Systems Biology, German Center for Neurodegenerative Diseases, Göttingen, Germany 3 Theoretical Computer Science and Algorithmic Methods, Institute of Computer Science, Georg-August-University Göttingen, Germany § Corresponding authors # Current address: Roche Pharmaceutical Research and Early Development, Pharmaceutical Sciences, Roche Innovation Center Basel, F. Hoffmann-La Roche Ltd., Basel, Switzerland $ Current address: Research and Development - Data Management (RD-DM), KWS SAAT SE, Einbeck, Germany * These authors contributed equally E-mail addresses: KH: [email protected], RV: [email protected], RR: [email protected], DS: [email protected], BH: [email protected], OS: [email protected], SB: [email protected], MK: [email protected] - 1 - bioRxiv preprint doi: https://doi.org/10.1101/133215; this version posted May 2, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Development of Novel Analysis and Data Integration Systems to Understand Human Gene Regulation
Development of novel analysis and data integration systems to understand human gene regulation Dissertation zur Erlangung des Doktorgrades Dr. rer. nat. der Fakult¨atf¨urMathematik und Informatik der Georg-August-Universit¨atG¨ottingen im PhD Programme in Computer Science (PCS) der Georg-August University School of Science (GAUSS) vorgelegt von Raza-Ur Rahman aus Pakistan G¨ottingen,April 2018 Prof. Dr. Stefan Bonn, Zentrum f¨urMolekulare Neurobiologie (ZMNH), Betreuungsausschuss: Institut f¨urMedizinische Systembiologie, Hamburg Prof. Dr. Tim Beißbarth, Institut f¨urMedizinische Statistik, Universit¨atsmedizin, Georg-August Universit¨at,G¨ottingen Prof. Dr. Burkhard Morgenstern, Institut f¨urMikrobiologie und Genetik Abtl. Bioinformatik, Georg-August Universit¨at,G¨ottingen Pr¨ufungskommission: Prof. Dr. Stefan Bonn, Zentrum f¨urMolekulare Neurobiologie (ZMNH), Referent: Institut f¨urMedizinische Systembiologie, Hamburg Prof. Dr. Tim Beißbarth, Institut f¨urMedizinische Statistik, Universit¨atsmedizin, Korreferent: Georg-August Universit¨at,G¨ottingen Prof. Dr. Burkhard Morgenstern, Weitere Mitglieder Institut f¨urMikrobiologie und Genetik Abtl. Bioinformatik, der Pr¨ufungskommission: Georg-August Universit¨at,G¨ottingen Prof. Dr. Carsten Damm, Institut f¨urInformatik, Georg-August Universit¨at,G¨ottingen Prof. Dr. Florentin W¨org¨otter, Physikalisches Institut Biophysik, Georg-August-Universit¨at,G¨ottingen Prof. Dr. Stephan Waack, Institut f¨urInformatik, Georg-August Universit¨at,G¨ottingen Tag der m¨undlichen Pr¨ufung: der 30. M¨arz2018 -

A Network Medicine Approach for Drug Repurposing in Duchenne Muscular Dystrophy
G C A T T A C G G C A T genes Article A Network Medicine Approach for Drug Repurposing in Duchenne Muscular Dystrophy Salvo Danilo Lombardo 1 , Maria Sofia Basile 2 , Rosella Ciurleo 2, Alessia Bramanti 2, Antonio Arcidiacono 3, Katia Mangano 3 , Placido Bramanti 2, Ferdinando Nicoletti 3,* and Paolo Fagone 3 1 Department of Structural & Computational Biology at the Max Perutz Labs, University of Vienna, 1010 Vienna, Austria; [email protected] 2 IRCCS Centro Neurolesi “Bonino-Pulejo”, Via Provinciale Palermo, Contrada Casazza, 98124 Messina, Italy; sofi[email protected] (M.S.B.); [email protected] (R.C.); [email protected] (A.B.); [email protected] (P.B.) 3 Department of Biomedical and Biotechnological Sciences, University of Catania, Via S. Sofia 89, 95123 Catania, Italy; [email protected] (A.A.); [email protected] (K.M.); [email protected] (P.F.) * Correspondence: [email protected] Abstract: Duchenne muscular dystrophy (DMD) is a progressive hereditary muscular disease caused by a lack of dystrophin, leading to membrane instability, cell damage, and inflammatory response. However, gene-editing alone is not enough to restore the healthy phenotype and additional treat- ments are required. In the present study, we have first conducted a meta-analysis of three microarray datasets, GSE38417, GSE3307, and GSE6011, to identify the differentially expressed genes (DEGs) be- tween healthy donors and DMD patients. We have then integrated this analysis with the knowledge Citation: Lombardo, S.D.; Basile, obtained from DisGeNET and DIAMOnD, a well-known algorithm for drug–gene association discov- M.S.; Ciurleo, R.; Bramanti, A.; eries in the human interactome. -

University of Groningen on the Elucidation of a Tumour Suppressor
University of Groningen On the elucidation of a tumour suppressor role of 3p in lung cancer Elst, Arja ter IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2006 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Elst, A. T. (2006). On the elucidation of a tumour suppressor role of 3p in lung cancer. s.n. Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 27-09-2021 Chapter 1 Candidate lung tumour suppressor regions at the short arm of chromosome 3. What evidence is there? Arja ter Elst Charles H.C.M. Buys Department of Medical Genetics, University Medical Center Groningen, Groningen, The Netherlands LUNG CANCER AND THE SHORT ARM OF CHROMOSOME 3 Lung cancer is the leading cause of cancer death among both men and women in the western world. -

Changes in the Gene Expression Profile During Spontaneous Migraine Attacks
www.nature.com/scientificreports OPEN Changes in the gene expression profle during spontaneous migraine attacks Lisette J. A. Kogelman1,7*, Katrine Falkenberg1,7, Alfonso Buil2, Pau Erola3, Julie Courraud4, Susan Svane Laursen4, Tom Michoel5, Jes Olesen1 & Thomas F. Hansen1,2,6* Migraine attacks are delimited, allowing investigation of changes during and outside attack. Gene expression fuctuates according to environmental and endogenous events and therefore, we hypothesized that changes in RNA expression during and outside a spontaneous migraine attack exist which are specifc to migraine. Twenty-seven migraine patients were assessed during a spontaneous migraine attack, including headache characteristics and treatment efect. Blood samples were taken during attack, two hours after treatment, on a headache-free day and after a cold pressor test. RNA- Sequencing, genotyping, and steroid profling were performed. RNA-Sequences were analyzed at gene level (diferential expression analysis) and at network level, and genomic and transcriptomic data were integrated. We found 29 diferentially expressed genes between ‘attack’ and ‘after treatment’, after subtracting non-migraine specifc genes, that were functioning in fatty acid oxidation, signaling pathways and immune-related pathways. Network analysis revealed mechanisms afected by changes in gene interactions, e.g. ‘ion transmembrane transport’. Integration of genomic and transcriptomic data revealed pathways related to sumatriptan treatment, i.e. ‘5HT1 type receptor mediated signaling pathway’. In conclusion, we uniquely investigated intra-individual changes in gene expression during a migraine attack. We revealed both genes and pathways potentially involved in the pathophysiology of migraine and/or migraine treatment. With a world-wide prevalence of 14.4% and global estimates of 5.6 years lost to disability, migraine is placed as the 2nd most disabling disease by the World Health Organization 1. -

Inter-Species Differences in Response to Hypoxia in Ipsc-Derived Cardiomyocytes from Humans and Chimpanzees
bioRxiv preprint doi: https://doi.org/10.1101/382895; this version posted August 2, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Inter-species differences in response to hypoxia in iPSC-derived cardiomyocytes from humans and chimpanzees Michelle C. Ward1* and Yoav Gilad1,2* 1Department of Medicine, University of Chicago, Chicago, IL, USA 2Department of Human Genetics, University of Chicago, Chicago, IL, USA *Correspondence should be addressed to M.C.W ([email protected]), and Y.G ([email protected]). 1 bioRxiv preprint doi: https://doi.org/10.1101/382895; this version posted August 2, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Despite anatomical similarities, there appear to be differences in susceptibility to cardiovascular disease between primates. For example, humans are prone to ischemia-induced myocardial infarction unlike chimpanzees, which tend to suffer from fibrotic disease. However, it is challenging to determine the relative contributions of genetic and environmental effects to complex disease phenotypes within and between primates. The ability to differentiate cardiomyocytes from induced pluripotent stem cells (iPSCs), now allows for direct inter-species comparisons of the gene regulatory response to disease-relevant perturbations. A consequence of ischemia is oxygen deprivation. -

SASH1, a New Potential Link Between Smoking and Atherosclerosis Henri Weidmann
SASH1, a new potential link between smoking and atherosclerosis Henri Weidmann To cite this version: Henri Weidmann. SASH1, a new potential link between smoking and atherosclerosis. Cardiology and cardiovascular system. Université Pierre et Marie Curie - Paris VI; Universität Hamburg, 2015. English. NNT : 2015PA066267. tel-01242976 HAL Id: tel-01242976 https://tel.archives-ouvertes.fr/tel-01242976 Submitted on 14 Dec 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Université Pierre et Marie Curie Ecole Doctorale 394: Physiology and physiopathology UMRS 1166 ICAN Institute, Insitute of Cardiometabolism And Nutrition Equipe 1: Genomics and physiopathology of cardiovascular diseases SASH1, a new potential link between smoking and atherosclerosis By Henri Weidmann To obtain the Degree of Doctor of physiology and physiopathology of the University Pierre et Marie Curie Directed by Dr Ewa Ninio Co-directed by Pr Tanja Zeller Publicly presented on the 23th of September 2015 Jury: Dr Mustapha Rouis, President of the Jury Dr Marie-Paul Jacob-Lenet, Reporter Dr Alain-Pierre Gadeau, Reporter Dr Klaus-Peter Janssen, Examiner Dr Fabienne Foufelle, Examiner 1 To my family To my friends 2 “Science is a way of thinking, much more than it is a body of knowledge” Carl Sagan 3 Acknowledgment Having reached the end of my thesis, I would like to thanks all the people that made this work possible and thus allowed me to attain my fondest dream, working in biological research. -

Differential Activity of the KRAS Oncogene by Method of Activation: Implications for Signaling and Therapeutic Intervention
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 12-2012 Differential Activity of the KRAS Oncogene by Method of Activation: Implications for Signaling and Therapeutic Intervention Nathan Ihle Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Medicine and Health Sciences Commons, Molecular Biology Commons, and the Systems Biology Commons Recommended Citation Ihle, Nathan, "Differential Activity of the KRAS Oncogene by Method of Activation: Implications for Signaling and Therapeutic Intervention" (2012). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 314. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/314 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. Differential Activity of the KRAS Oncogene by Method of Activation: Implications for Signaling and Therapeutic Intervention A DISSERTATION Presented to the Faculty of The University of Texas Health Science Center at Houston and The University of Texas M. D. Anderson Cancer Center Graduate School of Biomedical Sciences in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY By Nathan Ihle, B.S. -
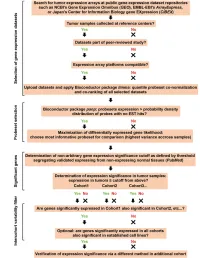
Supplementary Data
SUPPLEMENTAL INFORMATION A study restricted to chemokine receptors as well as a genome-wide transcript analysis uncovered CXCR4 as preferentially expressed in Ewing's sarcoma (Ewing's sarcoma) cells of metastatic origin (Figure 4). Transcriptome analyses showed that in addition to CXCR4, genes known to support cell motility and invasion topped the list of genes preferentially expressed in metastasis-derived cells (Figure 4D). These included kynurenine 3-monooxygenase (KMO), galectin-1 (LGALS1), gastrin-releasing peptide (GRP), procollagen C-endopeptidase enhancer (PCOLCE), and ephrin receptor B (EPHB3). KMO, a key enzyme of tryptophan catabolism, has not been linked to metastasis. Tryptophan and its catabolites, however, are involved in immune evasion by tumors, a process that can assist in tumor progression and metastasis (1). LGALS1, GRP, PCOLCE and EPHB3 have been linked to tumor progression and metastasis of several cancers (2-4). Top genes preferentially expressed in L-EDCL included genes that suppress cell motility and/or potentiate cell adhesion such as plakophilin 1 (PKP1), neuropeptide Y (NPY), or the metastasis suppressor TXNIP (5-7) (Figure 4D). Overall, L-EDCL were enriched in gene sets geared at optimizing nutrient transport and usage (Figure 4D; Supplementary Table 3), a state that may support the early stages of tumor growth. Once tumor growth outpaces nutrient and oxygen supplies, gene expression programs are usually switched to hypoxic response and neoangiogenesis, which ultimately lead to tumor egress and metastasis. Accordingly, gene sets involved in extracellular matrix remodeling, MAPK signaling, and response to hypoxia were up-regulated in M-EDCL (Figure 4D; Supplementary Table 4), consistent with their association to metastasis in other cancers (8, 9). -

Full Text (PDF)
medRxiv preprint doi: https://doi.org/10.1101/2020.12.29.20248986; this version posted January 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . Insights into the molecular mechanism of anticancer drug ruxolitinib repurposable in COVID-19 therapy Manisha Mandal Department of Physiology, MGM Medical College, Kishanganj-855107, India Email: [email protected], ORCID: https://orcid.org/0000-0002-9562-5534 Shyamapada Mandal* Department of Zoology, University of Gour Banga, Malda-732103, India Email: [email protected], ORCID: https://orcid.org/0000-0002-9488-3523 *Corresponding author: Email: [email protected]; [email protected] Abstract Due to non-availability of specific therapeutics against COVID-19, repurposing of approved drugs is a reasonable option. Cytokines imbalance in COVID-19 resembles cancer; exploration of anti-inflammatory agents, might reduce COVID-19 mortality. The current study investigates the effect of ruxolitinib treatment in SARS-CoV-2 infected alveolar cells compared to the uninfected one from the GSE5147507 dataset. The protein-protein interaction network, biological process and functional enrichment of differentially expressed genes were studied using STRING App of the Cytoscape software and R programming tools. The present study indicated that ruxolitinib treatment elicited similar response equivalent to that of SARS-CoV-2 uninfected situation by inducing defense response in host against virus infection by RLR and NOD like receptor pathways. Further, the effect of ruxolitinib in SARS- CoV-2 infection was mainly caused by significant suppression of IFIH1, IRF7 and MX1 genes as well as inhibition of DDX58/IFIH1-mediated induction of interferon- I and -II signalling. -

Genome-Wide Prediction of Dnase I Hypersensitivity Using Gene Expression
ARTICLE DOI: 10.1038/s41467-017-01188-x OPEN Genome-wide prediction of DNase I hypersensitivity using gene expression Weiqiang Zhou 1, Ben Sherwood1,3, Zhicheng Ji1, Yingchao Xue2, Fang Du1, Jiawei Bai1, Mingyao Ying2 & Hongkai Ji 1 We evaluate the feasibility of using a biological sample’s transcriptome to predict its genome- wide regulatory element activities measured by DNase I hypersensitivity (DH). We develop BIRD, Big Data Regression for predicting DH, to handle this high-dimensional problem. Applying BIRD to the Encyclopedia of DNA Elements (ENCODE) data, we found that to a large extent gene expression predicts DH, and information useful for prediction is contained in the whole transcriptome rather than limited to a regulatory element’s neighboring genes. We show applications of BIRD-predicted DH in predicting transcription factor-binding sites (TFBSs), turning publicly available gene expression samples in Gene Expression Omnibus (GEO) into a regulome database, predicting differential regulatory element activities, and facilitating regulome data analyses by serving as pseudo-replicates. Besides improving our understanding of the regulome–transcriptome relationship, this study suggests that transcriptome-based prediction can provide a useful new approach for regulome mapping. 1 Department of Biostatistics, Johns Hopkins University Bloomberg School of Public Health, 615 North Wolfe Street, Baltimore, MD 21205, USA. 2 Department of Neurology, Hugo W. Moser Research Institute at Kennedy Krieger and Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA. 3Present address: School of Business, University of Kansas, 1654 Naismith Drive, Lawrence, KS 66045, USA. Correspondence and requests for materials should be addressed to H.J. (email: [email protected]) NATURE COMMUNICATIONS | 8: 1038 | DOI: 10.1038/s41467-017-01188-x | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/s41467-017-01188-x fundamental question in functional genomics is how data analyses.