Disease Mutations in the Ryanodine Receptor N-Terminal Region Couple to a Mobile Intersubunit Interface
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Unnatural Verticilide Enantiomer Inhibits Type 2 Ryanodine Receptor-Mediated Calcium Leak and Is Antiarrhythmic
Unnatural verticilide enantiomer inhibits type 2 ryanodine receptor-mediated calcium leak and is antiarrhythmic Suzanne M. Batistea,1, Daniel J. Blackwellb,1, Kyungsoo Kimb,1, Dmytro O. Kryshtalb, Nieves Gomez-Hurtadob, Robyn T. Rebbeckc, Razvan L. Corneac, Jeffrey N. Johnstona,2, and Bjorn C. Knollmannb,2 aDepartment of Chemistry, Vanderbilt University, Nashville, TN 37235; bDepartment of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232; and cDepartment of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, MN 55455 Edited by Dale L. Boger, The Scripps Research Institute, La Jolla, CA, and approved January 15, 2019 (received for review September 27, 2018) Ca2+ leak via ryanodine receptor type 2 (RyR2) can cause poten- heart diseases associated with both atrial and ventricular arrhyth- tially fatal arrhythmias in a variety of heart diseases and has also mia (9). Mutations in RyR2 and its binding partners, which increase + been implicated in neurodegenerative and seizure disorders, mak- SR Ca2 leak, cause primary atrial and ventricular arrhythmia ing RyR2 an attractive therapeutic target for drug development. syndromes such as catecholaminergic polymorphic ventricular Here we synthesized and investigated the fungal natural product tachycardia (CPVT), providing strong evidence for the mechanistic and known insect RyR antagonist (−)-verticilide and several conge- contribution of RyR2 to arrhythmia risk in humans (10). Further ners to determine their activity against mammalian RyR2. Although support comes from gene-targeted mouse models of CPVT, where + the cyclooligomeric depsipeptide natural product (−)-verticilide had catecholamine-induced spontaneous Ca2 release from the SR no effect, its nonnatural enantiomer [ent-(+)-verticilide] signifi- via RyR2 generates potentially fatal cardiac arrhythmias (11, 12). -

Aquaporin Channels in the Heart—Physiology and Pathophysiology
International Journal of Molecular Sciences Review Aquaporin Channels in the Heart—Physiology and Pathophysiology Arie O. Verkerk 1,2,* , Elisabeth M. Lodder 2 and Ronald Wilders 1 1 Department of Medical Biology, Amsterdam University Medical Centers, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; [email protected] 2 Department of Experimental Cardiology, Amsterdam University Medical Centers, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +31-20-5664670 Received: 29 March 2019; Accepted: 23 April 2019; Published: 25 April 2019 Abstract: Mammalian aquaporins (AQPs) are transmembrane channels expressed in a large variety of cells and tissues throughout the body. They are known as water channels, but they also facilitate the transport of small solutes, gasses, and monovalent cations. To date, 13 different AQPs, encoded by the genes AQP0–AQP12, have been identified in mammals, which regulate various important biological functions in kidney, brain, lung, digestive system, eye, and skin. Consequently, dysfunction of AQPs is involved in a wide variety of disorders. AQPs are also present in the heart, even with a specific distribution pattern in cardiomyocytes, but whether their presence is essential for proper (electro)physiological cardiac function has not intensively been studied. This review summarizes recent findings and highlights the involvement of AQPs in normal and pathological cardiac function. We conclude that AQPs are at least implicated in proper cardiac water homeostasis and energy balance as well as heart failure and arsenic cardiotoxicity. However, this review also demonstrates that many effects of cardiac AQPs, especially on excitation-contraction coupling processes, are virtually unexplored. -

Supplemental Material
Supplemental Table B ARGs in alphabetical order Symbol Title 3 months 6 months 9 months 12 months 23 months ANOVA Direction Category 38597 septin 2 1557 ± 44 1555 ± 44 1579 ± 56 1655 ± 26 1691 ± 31 0.05219 up Intermediate 0610031j06rik kidney predominant protein NCU-G1 491 ± 6 504 ± 14 503 ± 11 527 ± 13 534 ± 12 0.04747 up Early Adult 1G5 vesicle-associated calmodulin-binding protein 662 ± 23 675 ± 17 629 ± 16 617 ± 20 583 ± 26 0.03129 down Intermediate A2m alpha-2-macroglobulin 262 ± 7 272 ± 8 244 ± 6 290 ± 7 353 ± 16 0.00000 up Midlife Aadat aminoadipate aminotransferase (synonym Kat2) 180 ± 5 201 ± 12 223 ± 7 244 ± 14 275 ± 7 0.00000 up Early Adult Abca2 ATP-binding cassette, sub-family A (ABC1), member 2 958 ± 28 1052 ± 58 1086 ± 36 1071 ± 44 1141 ± 41 0.05371 up Early Adult Abcb1a ATP-binding cassette, sub-family B (MDR/TAP), member 1A 136 ± 8 147 ± 6 147 ± 13 155 ± 9 185 ± 13 0.01272 up Midlife Acadl acetyl-Coenzyme A dehydrogenase, long-chain 423 ± 7 456 ± 11 478 ± 14 486 ± 13 512 ± 11 0.00003 up Early Adult Acadvl acyl-Coenzyme A dehydrogenase, very long chain 426 ± 14 414 ± 10 404 ± 13 411 ± 15 461 ± 10 0.01017 up Late Accn1 amiloride-sensitive cation channel 1, neuronal (degenerin) 242 ± 10 250 ± 9 237 ± 11 247 ± 14 212 ± 8 0.04972 down Late Actb actin, beta 12965 ± 310 13382 ± 170 13145 ± 273 13739 ± 303 14187 ± 269 0.01195 up Midlife Acvrinp1 activin receptor interacting protein 1 304 ± 18 285 ± 21 274 ± 13 297 ± 21 341 ± 14 0.03610 up Late Adk adenosine kinase 1828 ± 43 1920 ± 38 1922 ± 22 2048 ± 30 1949 ± 44 0.00797 up Early -
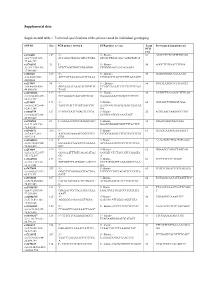
Supplemental Data Supplemental Table 1. Technical Specifications of the Primers Used for Individual Genotyping
Supplemental data Supplemental table 1. Technical specifications of the primers used for individual genotyping SNP ID Size PCR primer forward PCR primer reverse Temp Pyrosequencing primer(s) PCR (°C) rs776108 127 5’- 5’- Biotin- 61 ATACCTCTCATTTTGCAG chr3:77,825,927- ACCAGGCTAGGCATGCTATA GTCACTTAACAGCAGTGTGTCA 77,826,729 rs3746192 92 5’- 5’- Biotin- 56 AGGCTTGTAACCTGGA chr19:17,946,368- ATTCTAGGTGGCATGAGGG CTGGGGAGCAACAGAAGA 17,946,488 rs988147 169 5' - 5' - Biotine- 53 GCAGGGGGTAGAAATG chr6:45282108- AGCCATTAAAGAATTTCAAA TTGGATTTTATTCTTGTAATAGG 45282608 rs227849 94 5' - 5' - Biotine- 56 GGTTTAAGGTCTTTGCAT chr6:44,806,436- AGGAAAATAAACATGTGGTT TCTACCAATATTTTCTTTCGTAG 44,806,936 TAAG T rs10733833 127 5' - 5' - Biotine- 58 CATGTTTAAAACCTTTCAG chr10:68,418,227- GCCAAAACCAACAGTTCAT GAAAAAAATTGCACCTGTCTC 68,418,727 rs322609 197 5’- 5’-Biotin- 60 GGTAGCTGTGGGTGGA chr16:62,432,604- TAGTTGATTTTGCCAACCTG AAATGGGTGACAGAAGTAATAA 62,433,104 GA rs1884779 127 5’-TGGCTATTGGAGTTCTCA 5’-Biotin- 55 AGTGAATTAAGGGCTTGT chr20:45,857,969- CCATCCATCCCAAATAGT 45,858,469 rs4703908 83 5’- GAAAATGCCCAAGGTGAC 5’-Biotin- 52 GTGACAGTGGGCAAA chr5:71,802,353- GAATGTGGGTGTGTTTTACTCT 71,802,853 rs6946871 290 5’- 5’-Biotin- 61 GGAGGAAAGGAAAAGTT chr7:4,037,310- ATCAGATAAAATCGGCTTCT TCGGGAAGGTTTTTGTACTTTTG 4,037,810 GTG rs11865033 123 5’- 5’-Biotin- 61 AAAGTCTCTTCCTATGAGC chr16:78,082,945- AATAAACCAAGCCCTGAAAA ACTAAAATCCCCCTTTCCTCCA 78,083,445 GTC rs247004 170 5’- 5’-Biotin- 62 GGAAGCCAGACTAGCAG chr5:131,372,007- GGGGAATTTGTCAGAGATAG GGGATCCTCTACCATCCAAATA 131,372,507 GG -

THE NON-DYSTROPHIC MYOPATHIES JOHN PEARCE, M.B., M.R.C.P., Department of Neurology, the General Infirmary, Leeds
Postgrad Med J: first published as 10.1136/pgmj.41.476.347 on 1 June 1965. Downloaded from POSTGRAD. MED. J. (1965), 41, 347 THE NON-DYSTROPHIC MYOPATHIES JOHN PEARCE, M.B., M.R.C.P., Department of Neurology, The General Infirmary, Leeds. THE TERM 'myopathy' is applied to any disorder Polymyositis may affect people of any age, of the muscle fibre, the muscle fibre membrane, but the age of onset is from 30 to 60 in 60% the myoneural junction, or the muscle connect- of cases. The chief symptom is weakness of ive tissue. 'Non-dystrophic myopathy' includes proximal muscles of the arms and/or legs all diseases of muscle excluding those genetically in every case. Distal muscles are affected in determined primary degenerative myopathies, one third, and the neck muscles in two thirds collectively known as Muscular Dystrophy. of patients. Fever, muscular pain and tenderness TABLE 1 are seen most often in the more acute forms, CLASSIFICATION OF NON-DYSTROPHIC MYOPATHIES and their absence should not lead to neglecting 1. Inflammatory Polymyositis as a Myopathy Connective tissue disorders polymyositis possible diagnosis. Other inflammatory mvopathies Acute polymyositis is not common, but may 2. Metabolic Familial periodic paralysis progress rapidly and involve the respiratory Myopathy Muscle glycogenoses muscles, sometimes with a fatal termination Myoglobinuric myopathies within a few weeks or months. Myopathies associated with Subacute and electrolyte imbalance chronic forms are more frequent, and present Unclassified myopathies with a progressive weakness and moderate of shoulder 3. Endocrine Thyrotoxicosis wasting and pelvic girdle muscles. Myopathy Cushing's Syndrome There is sometimes no systemic disturbance, Steroid myopathy and pain and tenderness are lacking. -

Ion Channels 3 1
r r r Cell Signalling Biology Michael J. Berridge Module 3 Ion Channels 3 1 Module 3 Ion Channels Synopsis Ion channels have two main signalling functions: either they can generate second messengers or they can function as effectors by responding to such messengers. Their role in signal generation is mainly centred on the Ca2 + signalling pathway, which has a large number of Ca2+ entry channels and internal Ca2+ release channels, both of which contribute to the generation of Ca2 + signals. Ion channels are also important effectors in that they mediate the action of different intracellular signalling pathways. There are a large number of K+ channels and many of these function in different + aspects of cell signalling. The voltage-dependent K (KV) channels regulate membrane potential and + excitability. The inward rectifier K (Kir) channel family has a number of important groups of channels + + such as the G protein-gated inward rectifier K (GIRK) channels and the ATP-sensitive K (KATP) + + channels. The two-pore domain K (K2P) channels are responsible for the large background K current. Some of the actions of Ca2 + are carried out by Ca2+-sensitive K+ channels and Ca2+-sensitive Cl − channels. The latter are members of a large group of chloride channels and transporters with multiple functions. There is a large family of ATP-binding cassette (ABC) transporters some of which have a signalling role in that they extrude signalling components from the cell. One of the ABC transporters is the cystic − − fibrosis transmembrane conductance regulator (CFTR) that conducts anions (Cl and HCO3 )and contributes to the osmotic gradient for the parallel flow of water in various transporting epithelia. -

Physiological and Pathophysiological Regulation of the Ryanodine Receptor in Skeletal Muscle
Physiological and pathophysiological regulation of the ryanodine receptor in skeletal muscle Alisa Umanskaya Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2015 © 2015 Alisa Umanskaya All rights reserved Abstract Physiological and pathophysiological regulation of ryanodine receptor in skeletal muscle Alisa Umanskaya Ryanodine receptor calcium release channels are essential for skeletal muscle contraction, as they mediate the release of calcium ions from intracellular stores into the cytosol. The data presented in this dissertation demonstrate the evolutionarily conserved mechanisms of skeletal muscle ryanodine receptor regulation in the physiological and pathophysiological states. Adrenergic stimulation causes increased skeletal muscle force, however, despite the well- established role of this physiological response, the molecular mechanism is not known. Here we present a mechanism whereby phosphorylation of a single amino acid on the ryanodine receptor is a key signal in the physiological stress-induced inotropic response in mouse skeletal muscle. Therefore acute post-translational modifications of ryanodine receptor channels are important for healthy muscle contraction. Conversely, chronic stress-induced post-translational modifications result in poorly functioning murine ryanodine receptor channels that contribute to skeletal muscle dysfunction in age- dependent skeletal muscle weakness and Muscular Dystrophies. Finally, we present data that demonstrates striking evolutionary conservation in ryanodine receptor regulation in the physiological and pathophysiological states between mice and C. elegans. This work has broad implications for understanding the underlying mechanisms of skeletal muscle contraction and important disorders that affect human health. Furthermore, this works presents ryanodine receptor channels as a viable therapeutic target for age-related skeletal muscle weakness, Muscular Dystrophies, and also implicates C. -

Spatial Distribution of Leading Pacemaker Sites in the Normal, Intact Rat Sinoa
Supplementary Material Supplementary Figure 1: Spatial distribution of leading pacemaker sites in the normal, intact rat sinoatrial 5 nodes (SAN) plotted along a normalized y-axis between the superior vena cava (SVC) and inferior vena 6 cava (IVC) and a scaled x-axis in millimeters (n = 8). Colors correspond to treatment condition (black: 7 baseline, blue: 100 µM Acetylcholine (ACh), red: 500 nM Isoproterenol (ISO)). 1 Supplementary Figure 2: Spatial distribution of leading pacemaker sites before and after surgical 3 separation of the rat SAN (n = 5). Top: Intact SAN preparations with leading pacemaker sites plotted during 4 baseline conditions. Bottom: Surgically cut SAN preparations with leading pacemaker sites plotted during 5 baseline conditions (black) and exposure to pharmacological stimulation (blue: 100 µM ACh, red: 500 nM 6 ISO). 2 a &DUGLDFIoQChDQQHOV .FQM FOXVWHU &DFQDG &DFQDK *MD &DFQJ .FQLS .FQG .FQK .FQM &DFQDF &DFQE .FQM í $WSD .FQD .FQM í .FQN &DVT 5\U .FQM &DFQJ &DFQDG ,WSU 6FQD &DFQDG .FQQ &DFQDJ &DFQDG .FQD .FQT 6FQD 3OQ 6FQD +FQ *MD ,WSU 6FQE +FQ *MG .FQN .FQQ .FQN .FQD .FQE .FQQ +FQ &DFQDD &DFQE &DOP .FQM .FQD .FQN .FQG .FQN &DOP 6FQD .FQD 6FQE 6FQD 6FQD ,WSU +FQ 6FQD 5\U 6FQD 6FQE 6FQD .FQQ .FQH 6FQD &DFQE 6FQE .FQM FOXVWHU V6$1 L6$1 5$ /$ 3 b &DUGLDFReFHSWRUV $GUDF FOXVWHU $GUDD &DY &KUQE &KUP &KJD 0\O 3GHG &KUQD $GUE $GUDG &KUQE 5JV í 9LS $GUDE 7SP í 5JV 7QQF 3GHE 0\K $GUE *QDL $QN $GUDD $QN $QN &KUP $GUDE $NDS $WSE 5DPS &KUP 0\O &KUQD 6UF &KUQH $GUE &KUQD FOXVWHU V6$1 L6$1 5$ /$ 4 c 1HXURQDOPURWHLQV -

Central Core Disease - a Case Report
The Korean Journal of Pathology 2004; 38: 68-71 Central Core Disease - A Case Report - Ji Hoon Kim∙Young S. Park Central core disease is a rare autosomal dominantly inherited non-progressive congenital myopa- Sung-Hye Park∙Je G. Chi thy, which is pathologically characterized by the formation of a ‘‘core’’. We report a 28-year-old female with non-progressive muscle weakness, who had a hypotonic posture at birth. The devel- Department of Pathology, Seoul National opmental milestones were delayed with her first walking at 18 months of age. She could not University College of Medicine, Seoul, run or walk a long distance and weight-bearing tasks were almost impossible. None of her Korea family members showed motor symptoms. An investigation of the electromyography and nerve conduction velocity showed non-specific results. A gastrocnemius muscle biopsy revealed Received : November 14, 2003 central cores in approximately 70% of myofibers with a type 1 myofiber predominance and Accepted : January 7, 2004 deranged sarcolemmal structures. To the best of our knowledge, this is the fifth report of cen- tral core disease in the Korean literature. Corresponding Author Sung-Hye Park, M.D. Department of Pathology, Seoul National University College of Medicine, 28 Yongon-dong, Chongno-gu, Seoul 110-799, Korea Tel: 02-740-8278 Fax: 02-765-5600 E-mail: [email protected] Key Words : Myopathy, Central Core-Muscle, Skeletal-Ultrastructure Central core disease is a form of congenital non-progressive right upper extremity. The symptoms were relieved with the help myopathy, which was first described by Shy and Magee in 1956. -

RYR2 Gene Ryanodine Receptor 2
RYR2 gene ryanodine receptor 2 Normal Function The RYR2 gene provides instructions for making a protein called ryanodine receptor 2. This protein is part of a family of ryanodine receptors, which form channels that transport positively charged calcium atoms (calcium ions) within cells. Channels made with the ryanodine receptor 2 protein are found in heart (cardiac) muscle cells called myocytes. These channels are embedded in the outer membrane of a cell structure called the sarcoplasmic reticulum, which acts as a storage center for calcium ions. The RYR2 channel controls the flow of calcium ions out of the sarcoplasmic reticulum. For the heart to beat normally, the cardiac muscle must tense (contract) and relax in a coordinated way. This cycle of muscle contraction and relaxation results from the precise control of calcium ions within myocytes. In response to certain signals, the RYR2 channel releases calcium ions from the sarcoplasmic reticulum into the surrounding cell fluid (the cytoplasm). The resulting increase in calcium ion concentration triggers the cardiac muscle to contract, which pumps blood out of the heart. Calcium ions are then transported back into the sarcoplasmic reticulum, and the cardiac muscle relaxes. In this way, the release and reuptake of calcium ions in myocytes produces a regular heart rhythm. Health Conditions Related to Genetic Changes Catecholaminergic polymorphic ventricular tachycardia More than 200 mutations in the RYR2 gene have been found to cause catecholaminergic polymorphic ventricular tachycardia (CPVT), a heart condition characterized by an abnormal heart rhythm (arrhythmia) during exercise or emotional stress, which can be fatal. Almost all of the RYR2 gene mutations involved in CPVT change single protein building blocks (amino acids) in the ryanodine receptor 2 protein. -

Central Core Disease in Focus
Central Core Disease In Focus Fast Facts Central core disease (CCD) is a genetic muscle disease characterized by the appearance of corelike struc- tures running through the centers of muscle fibers. The cores are areas of metabolic inactivity. Symptoms vary widely in sever- ity and can begin anywhere from infancy to adulthood. They include weakness, muscle cramps, orthope- dic abnormalities (spinal curvature, foot deformities, hip dislocations) caused by the weakness, and a dan- gerous susceptibility to malignant hyperthermia, an adverse reaction to anesthesia. The disease is usually dominantly In Focus inherited, meaning only one gene mutation, inherited from one parent, CCD: A Disease with Many Faces is necessary to cause symptoms. by Margaret Wahl The underlying molecular cause of the disease is an abnormality of orty-one-year-old Sandy Doak remem- since Doak is 5 feet tall and weighs 128 calcium release from deep inside Fbers that she was never athletic, pounds, the test result said her body was the muscle fibers. Normally, a signal couldn’t do sit-ups, and always had trou- 38 percent fat. There was only one way from a nerve fiber tells a muscle fiber ble finishing physical tasks. But, she says, that could be the case, the fitness expert to contract and, after a cascade of “I never really thought I had anything and she agreed: She had very little mus- events, calcium is released in a burst wrong. I just thought I was awkward. I cle. For Doak, it was a clue that something from internal storage areas. Calcium knew I was weaker than others. -

Neurological Diseases Caused by Ion-Channel Mutations Frank Weinreich and Thomas J Jentsch*
409 Neurological diseases caused by ion-channel mutations Frank Weinreich and Thomas J Jentsch* During the past decade, mutations in several ion-channel humans. This is probably the case for important Na+-chan- genes have been shown to cause inherited neurological nel isoforms, such as those dominating excitation in diseases. This is not surprising given the large number of skeletal muscle or heart, and may also be the case for the different ion channels and their prominent role in signal two channel subunits that assemble to form M-type processing. Biophysical studies of mutant ion channels in vitro K+-channels, which are key regulators of neuronal allow detailed investigations of the basic mechanism excitability [8••,9•]. This concept is supported by the underlying these ‘channelopathies’. A full understanding of observation that many channelopathies are paroxysmal these diseases, however, requires knowing the roles these (i.e. cause transient convulsions): mutations leading to a channels play in their cellular and systemic context. Differences constant disability might be incompatible with life, or may in this context often cause different phenotypes in humans and significantly decrease the frequency of the mutation with- mice. The situation is further complicated by the developmental in the human population. In contrast to the severe effects and other secondary effects that might result from ion- symptoms associated with the loss of function of certain channel mutations. Recent studies have described the different key ion channels, the large number of ion-channel isoforms thresholds to which ion-channel function must be decreased in may lead to a functional redundancy under most circum- order to cause disease.