Physiological and Pathophysiological Regulation of the Ryanodine Receptor in Skeletal Muscle
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hearing Aging Is 14.1±0.4% GWAS-Heritable
medRxiv preprint doi: https://doi.org/10.1101/2021.07.05.21260048; this version posted July 7, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC 4.0 International license . Predicting age from hearing test results with machine learning reveals the genetic and environmental factors underlying accelerated auditory aging Alan Le Goallec1,2+, Samuel Diai1+, Théo Vincent1, Chirag J. Patel1* 1Department of Biomedical Informatics, Harvard Medical School, Boston, MA, 02115, USA 2Department of Systems, Synthetic and Quantitative Biology, Harvard University, Cambridge, MA, 02118, USA +Co-first authors *Corresponding author Contact information: Chirag J Patel [email protected] Abstract With the aging of the world population, age-related hearing loss (presbycusis) and other hearing disorders such as tinnitus become more prevalent, leading to reduced quality of life and social isolation. Unveiling the genetic and environmental factors leading to age-related auditory disorders could suggest lifestyle and therapeutic interventions to slow auditory aging. In the following, we built the first machine learning-based hearing age predictor by training models to predict chronological age from hearing test results (root mean squared error=7.10±0.07 years; R-Squared=31.4±0.8%). We defined hearing age as the prediction outputted by the model on unseen samples, and accelerated auditory aging as the difference between a participant’s hearing age and age. We then performed a genome wide association study [GWAS] and found that accelerated hearing aging is 14.1±0.4% GWAS-heritable. -

Unnatural Verticilide Enantiomer Inhibits Type 2 Ryanodine Receptor-Mediated Calcium Leak and Is Antiarrhythmic
Unnatural verticilide enantiomer inhibits type 2 ryanodine receptor-mediated calcium leak and is antiarrhythmic Suzanne M. Batistea,1, Daniel J. Blackwellb,1, Kyungsoo Kimb,1, Dmytro O. Kryshtalb, Nieves Gomez-Hurtadob, Robyn T. Rebbeckc, Razvan L. Corneac, Jeffrey N. Johnstona,2, and Bjorn C. Knollmannb,2 aDepartment of Chemistry, Vanderbilt University, Nashville, TN 37235; bDepartment of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232; and cDepartment of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, MN 55455 Edited by Dale L. Boger, The Scripps Research Institute, La Jolla, CA, and approved January 15, 2019 (received for review September 27, 2018) Ca2+ leak via ryanodine receptor type 2 (RyR2) can cause poten- heart diseases associated with both atrial and ventricular arrhyth- tially fatal arrhythmias in a variety of heart diseases and has also mia (9). Mutations in RyR2 and its binding partners, which increase + been implicated in neurodegenerative and seizure disorders, mak- SR Ca2 leak, cause primary atrial and ventricular arrhythmia ing RyR2 an attractive therapeutic target for drug development. syndromes such as catecholaminergic polymorphic ventricular Here we synthesized and investigated the fungal natural product tachycardia (CPVT), providing strong evidence for the mechanistic and known insect RyR antagonist (−)-verticilide and several conge- contribution of RyR2 to arrhythmia risk in humans (10). Further ners to determine their activity against mammalian RyR2. Although support comes from gene-targeted mouse models of CPVT, where + the cyclooligomeric depsipeptide natural product (−)-verticilide had catecholamine-induced spontaneous Ca2 release from the SR no effect, its nonnatural enantiomer [ent-(+)-verticilide] signifi- via RyR2 generates potentially fatal cardiac arrhythmias (11, 12). -

Aquaporin Channels in the Heart—Physiology and Pathophysiology
International Journal of Molecular Sciences Review Aquaporin Channels in the Heart—Physiology and Pathophysiology Arie O. Verkerk 1,2,* , Elisabeth M. Lodder 2 and Ronald Wilders 1 1 Department of Medical Biology, Amsterdam University Medical Centers, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; [email protected] 2 Department of Experimental Cardiology, Amsterdam University Medical Centers, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +31-20-5664670 Received: 29 March 2019; Accepted: 23 April 2019; Published: 25 April 2019 Abstract: Mammalian aquaporins (AQPs) are transmembrane channels expressed in a large variety of cells and tissues throughout the body. They are known as water channels, but they also facilitate the transport of small solutes, gasses, and monovalent cations. To date, 13 different AQPs, encoded by the genes AQP0–AQP12, have been identified in mammals, which regulate various important biological functions in kidney, brain, lung, digestive system, eye, and skin. Consequently, dysfunction of AQPs is involved in a wide variety of disorders. AQPs are also present in the heart, even with a specific distribution pattern in cardiomyocytes, but whether their presence is essential for proper (electro)physiological cardiac function has not intensively been studied. This review summarizes recent findings and highlights the involvement of AQPs in normal and pathological cardiac function. We conclude that AQPs are at least implicated in proper cardiac water homeostasis and energy balance as well as heart failure and arsenic cardiotoxicity. However, this review also demonstrates that many effects of cardiac AQPs, especially on excitation-contraction coupling processes, are virtually unexplored. -

Table 2. Significant
Table 2. Significant (Q < 0.05 and |d | > 0.5) transcripts from the meta-analysis Gene Chr Mb Gene Name Affy ProbeSet cDNA_IDs d HAP/LAP d HAP/LAP d d IS Average d Ztest P values Q-value Symbol ID (study #5) 1 2 STS B2m 2 122 beta-2 microglobulin 1452428_a_at AI848245 1.75334941 4 3.2 4 3.2316485 1.07398E-09 5.69E-08 Man2b1 8 84.4 mannosidase 2, alpha B1 1416340_a_at H4049B01 3.75722111 3.87309653 2.1 1.6 2.84852656 5.32443E-07 1.58E-05 1110032A03Rik 9 50.9 RIKEN cDNA 1110032A03 gene 1417211_a_at H4035E05 4 1.66015788 4 1.7 2.82772795 2.94266E-05 0.000527 NA 9 48.5 --- 1456111_at 3.43701477 1.85785922 4 2 2.8237185 9.97969E-08 3.48E-06 Scn4b 9 45.3 Sodium channel, type IV, beta 1434008_at AI844796 3.79536664 1.63774235 3.3 2.3 2.75319499 1.48057E-08 6.21E-07 polypeptide Gadd45gip1 8 84.1 RIKEN cDNA 2310040G17 gene 1417619_at 4 3.38875643 1.4 2 2.69163229 8.84279E-06 0.0001904 BC056474 15 12.1 Mus musculus cDNA clone 1424117_at H3030A06 3.95752801 2.42838452 1.9 2.2 2.62132809 1.3344E-08 5.66E-07 MGC:67360 IMAGE:6823629, complete cds NA 4 153 guanine nucleotide binding protein, 1454696_at -3.46081884 -4 -1.3 -1.6 -2.6026947 8.58458E-05 0.0012617 beta 1 Gnb1 4 153 guanine nucleotide binding protein, 1417432_a_at H3094D02 -3.13334396 -4 -1.6 -1.7 -2.5946297 1.04542E-05 0.0002202 beta 1 Gadd45gip1 8 84.1 RAD23a homolog (S. -

Supplemental Material
Supplemental Table B ARGs in alphabetical order Symbol Title 3 months 6 months 9 months 12 months 23 months ANOVA Direction Category 38597 septin 2 1557 ± 44 1555 ± 44 1579 ± 56 1655 ± 26 1691 ± 31 0.05219 up Intermediate 0610031j06rik kidney predominant protein NCU-G1 491 ± 6 504 ± 14 503 ± 11 527 ± 13 534 ± 12 0.04747 up Early Adult 1G5 vesicle-associated calmodulin-binding protein 662 ± 23 675 ± 17 629 ± 16 617 ± 20 583 ± 26 0.03129 down Intermediate A2m alpha-2-macroglobulin 262 ± 7 272 ± 8 244 ± 6 290 ± 7 353 ± 16 0.00000 up Midlife Aadat aminoadipate aminotransferase (synonym Kat2) 180 ± 5 201 ± 12 223 ± 7 244 ± 14 275 ± 7 0.00000 up Early Adult Abca2 ATP-binding cassette, sub-family A (ABC1), member 2 958 ± 28 1052 ± 58 1086 ± 36 1071 ± 44 1141 ± 41 0.05371 up Early Adult Abcb1a ATP-binding cassette, sub-family B (MDR/TAP), member 1A 136 ± 8 147 ± 6 147 ± 13 155 ± 9 185 ± 13 0.01272 up Midlife Acadl acetyl-Coenzyme A dehydrogenase, long-chain 423 ± 7 456 ± 11 478 ± 14 486 ± 13 512 ± 11 0.00003 up Early Adult Acadvl acyl-Coenzyme A dehydrogenase, very long chain 426 ± 14 414 ± 10 404 ± 13 411 ± 15 461 ± 10 0.01017 up Late Accn1 amiloride-sensitive cation channel 1, neuronal (degenerin) 242 ± 10 250 ± 9 237 ± 11 247 ± 14 212 ± 8 0.04972 down Late Actb actin, beta 12965 ± 310 13382 ± 170 13145 ± 273 13739 ± 303 14187 ± 269 0.01195 up Midlife Acvrinp1 activin receptor interacting protein 1 304 ± 18 285 ± 21 274 ± 13 297 ± 21 341 ± 14 0.03610 up Late Adk adenosine kinase 1828 ± 43 1920 ± 38 1922 ± 22 2048 ± 30 1949 ± 44 0.00797 up Early -
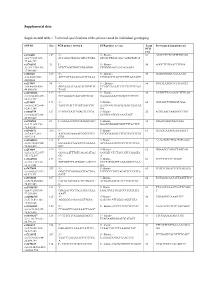
Supplemental Data Supplemental Table 1. Technical Specifications of the Primers Used for Individual Genotyping
Supplemental data Supplemental table 1. Technical specifications of the primers used for individual genotyping SNP ID Size PCR primer forward PCR primer reverse Temp Pyrosequencing primer(s) PCR (°C) rs776108 127 5’- 5’- Biotin- 61 ATACCTCTCATTTTGCAG chr3:77,825,927- ACCAGGCTAGGCATGCTATA GTCACTTAACAGCAGTGTGTCA 77,826,729 rs3746192 92 5’- 5’- Biotin- 56 AGGCTTGTAACCTGGA chr19:17,946,368- ATTCTAGGTGGCATGAGGG CTGGGGAGCAACAGAAGA 17,946,488 rs988147 169 5' - 5' - Biotine- 53 GCAGGGGGTAGAAATG chr6:45282108- AGCCATTAAAGAATTTCAAA TTGGATTTTATTCTTGTAATAGG 45282608 rs227849 94 5' - 5' - Biotine- 56 GGTTTAAGGTCTTTGCAT chr6:44,806,436- AGGAAAATAAACATGTGGTT TCTACCAATATTTTCTTTCGTAG 44,806,936 TAAG T rs10733833 127 5' - 5' - Biotine- 58 CATGTTTAAAACCTTTCAG chr10:68,418,227- GCCAAAACCAACAGTTCAT GAAAAAAATTGCACCTGTCTC 68,418,727 rs322609 197 5’- 5’-Biotin- 60 GGTAGCTGTGGGTGGA chr16:62,432,604- TAGTTGATTTTGCCAACCTG AAATGGGTGACAGAAGTAATAA 62,433,104 GA rs1884779 127 5’-TGGCTATTGGAGTTCTCA 5’-Biotin- 55 AGTGAATTAAGGGCTTGT chr20:45,857,969- CCATCCATCCCAAATAGT 45,858,469 rs4703908 83 5’- GAAAATGCCCAAGGTGAC 5’-Biotin- 52 GTGACAGTGGGCAAA chr5:71,802,353- GAATGTGGGTGTGTTTTACTCT 71,802,853 rs6946871 290 5’- 5’-Biotin- 61 GGAGGAAAGGAAAAGTT chr7:4,037,310- ATCAGATAAAATCGGCTTCT TCGGGAAGGTTTTTGTACTTTTG 4,037,810 GTG rs11865033 123 5’- 5’-Biotin- 61 AAAGTCTCTTCCTATGAGC chr16:78,082,945- AATAAACCAAGCCCTGAAAA ACTAAAATCCCCCTTTCCTCCA 78,083,445 GTC rs247004 170 5’- 5’-Biotin- 62 GGAAGCCAGACTAGCAG chr5:131,372,007- GGGGAATTTGTCAGAGATAG GGGATCCTCTACCATCCAAATA 131,372,507 GG -

Variants in the KCNE1 Or KCNE3 Gene and Risk of Ménière’S Disease: a Meta-Analysis
Journal of Vestibular Research 25 (2015) 211–218 211 DOI 10.3233/VES-160569 IOS Press Variants in the KCNE1 or KCNE3 gene and risk of Ménière’s disease: A meta-analysis Yuan-Jun Li, Zhan-Guo Jin and Xian-Rong Xu∗ The Center of Clinical Aviation Medicine, General Hospital of Air Force, Beijing, China Received 1 August 2015 Accepted 8 December 2015 Abstract. BACKGROUND: Ménière’s disease (MD) is defined as an idiopathic disorder of the inner ear characterized by the triad of tinnitus, vertigo, and sensorineural hearing loss. Although many studies have evaluated the association between variants in the KCNE1 or KCNE3 gene and MD risk, debates still exist. OBJECTIVE: Our aim is to evaluate the association between KCNE gene variants, including KCNE1 rs1805127 and KCNE3 rs2270676, and the risk of MD by a systematic review. METHODS: We searched the literature in PubMed, SCOPUS and EMBASE through May 2015. We calculated pooled odds ra- tios (OR) and 95% confidence intervals (CIs) using a fixed-effects model or a random-effects model for the risk to MD associated with different KCNE gene variants. The heterogeneity assumption decided the effect model. RESULTS: A total of three relevant studies, with 302 MD cases and 515 controls, were included in this meta-analysis. The results indicated that neither the KCNE1 rs1805127 variant (for G vs. A: OR = 0.724, 95%CI 0.320, 1.638, P = 0.438), nor the KCNE3 rs2270676 variant (for T vs. C: OR = 0.714, 95%CI 0.327, 1.559, P = 0.398) was associated with MD risk. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Figure S1. Representative Report Generated by the Ion Torrent System Server for Each of the KCC71 Panel Analysis and Pcafusion Analysis
Figure S1. Representative report generated by the Ion Torrent system server for each of the KCC71 panel analysis and PCaFusion analysis. (A) Details of the run summary report followed by the alignment summary report for the KCC71 panel analysis sequencing. (B) Details of the run summary report for the PCaFusion panel analysis. A Figure S1. Continued. Representative report generated by the Ion Torrent system server for each of the KCC71 panel analysis and PCaFusion analysis. (A) Details of the run summary report followed by the alignment summary report for the KCC71 panel analysis sequencing. (B) Details of the run summary report for the PCaFusion panel analysis. B Figure S2. Comparative analysis of the variant frequency found by the KCC71 panel and calculated from publicly available cBioPortal datasets. For each of the 71 genes in the KCC71 panel, the frequency of variants was calculated as the variant number found in the examined cases. Datasets marked with different colors and sample numbers of prostate cancer are presented in the upper right. *Significantly high in the present study. Figure S3. Seven subnetworks extracted from each of seven public prostate cancer gene networks in TCNG (Table SVI). Blue dots represent genes that include initial seed genes (parent nodes), and parent‑child and child‑grandchild genes in the network. Graphical representation of node‑to‑node associations and subnetwork structures that differed among and were unique to each of the seven subnetworks. TCNG, The Cancer Network Galaxy. Figure S4. REVIGO tree map showing the predicted biological processes of prostate cancer in the Japanese. Each rectangle represents a biological function in terms of a Gene Ontology (GO) term, with the size adjusted to represent the P‑value of the GO term in the underlying GO term database. -

Transcriptional Control of Tissue-Resident Memory T Cell Generation
Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2019 © 2019 Filip Cvetkovski All rights reserved ABSTRACT Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Tissue-resident memory T cells (TRM) are a non-circulating subset of memory that are maintained at sites of pathogen entry and mediate optimal protection against reinfection. Lung TRM can be generated in response to respiratory infection or vaccination, however, the molecular pathways involved in CD4+TRM establishment have not been defined. Here, we performed transcriptional profiling of influenza-specific lung CD4+TRM following influenza infection to identify pathways implicated in CD4+TRM generation and homeostasis. Lung CD4+TRM displayed a unique transcriptional profile distinct from spleen memory, including up-regulation of a gene network induced by the transcription factor IRF4, a known regulator of effector T cell differentiation. In addition, the gene expression profile of lung CD4+TRM was enriched in gene sets previously described in tissue-resident regulatory T cells. Up-regulation of immunomodulatory molecules such as CTLA-4, PD-1, and ICOS, suggested a potential regulatory role for CD4+TRM in tissues. Using loss-of-function genetic experiments in mice, we demonstrate that IRF4 is required for the generation of lung-localized pathogen-specific effector CD4+T cells during acute influenza infection. Influenza-specific IRF4−/− T cells failed to fully express CD44, and maintained high levels of CD62L compared to wild type, suggesting a defect in complete differentiation into lung-tropic effector T cells. -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

KCNE1 and KCNE3 Modulate KCNQ1 Channels by Affecting
KCNE1 and KCNE3 modulate KCNQ1 channels by PNAS PLUS affecting different gating transitions Rene Barro-Soriaa,1,2, Rosamary Ramentola, Sara I. Liina,3, Marta E. Pereza, Robert S. Kassb, and H. Peter Larssona,2 aDepartment of Physiology and Biophysics, Miller School of Medicine, University of Miami, Miami, FL 33136; and bDepartment of Pharmacology, College of Physicians and Surgeons, Columbia University, New York, NY 10032 Edited by Ramon Latorre, Universidad de Valparaíso, Valparaíso, Chile, and approved July 19, 2017 (received for review June 16, 2017) KCNE β-subunits assemble with and modulate the properties of on the activation of KCNQ1 channels is still debated. Here, we voltage-gated K+ channels. In the heart, KCNE1 associates with simultaneously track changes in voltage sensor movement and in the α-subunit KCNQ1 to generate the slowly activating, voltage- gate opening of KCNQ1/KCNE1 and KCNQ1/KCNE3 channels dependent potassium current (IKs) in the heart that controls the using voltage clamp fluorometry (VCF) to determine the differ- repolarization phase of cardiac action potentials. By contrast, in ences in the molecular mechanisms by which KCNE1 and KCNE3 epithelial cells from the colon, stomach, and kidney, KCNE3 coas- alter KCNQ1 channel gating. + sembles with KCNQ1 to form K channels that are voltage- Pore-forming KCNQ1 channel α-subunits form tetramers. Each + independent K channels in the physiological voltage range and α-subunit has six transmembrane (TM) segments (S1–S6) divided important for controlling water and salt secretion and absorption. into two functional domains: a voltage sensor domain comprising How KCNE1 and KCNE3 subunits modify KCNQ1 channel gating so four peripheral TM helices (S1–S4) and a centrally located pore differently is largely unknown.