MAPK Kinase 3 Is a Tumor Suppressor with Reduced Copy Number in Breast Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MAP2K3 (Human) Recombinant Protein (Q01)
MAP2K3 (Human) Recombinant phosphorylates and thus activates MAPK14/p38-MAPK. Protein (Q01) This kinase can be activated by insulin, and is necessary for the expression of glucose transporter. Expression of Catalog Number: H00005606-Q01 RAS oncogene is found to result in the accumulation of the active form of this kinase, which thus leads to the Regulation Status: For research use only (RUO) constitutive activation of MAPK14, and confers oncogenic transformation of primary cells. The inhibition Product Description: Human MAP2K3 partial ORF ( of this kinase is involved in the pathogenesis of Yersina AAH32478, 1 a.a. - 100 a.a.) recombinant protein with pseudotuberculosis. Multiple alternatively spliced GST-tag at N-terminal. transcript variants that encode distinct isoforms have been reported for this gene. [provided by RefSeq] Sequence: MESPASSQPASMPQSKGKSKRKKDLRISCMSKPPAP NPTPPRNLDSRTFITIGDRNFEVEADDLVTISELGRGAY GVVEKVRHAQSGTIMAVKRIRATVN Host: Wheat Germ (in vitro) Theoretical MW (kDa): 36.63 Applications: AP, Array, ELISA, WB-Re (See our web site product page for detailed applications information) Protocols: See our web site at http://www.abnova.com/support/protocols.asp or product page for detailed protocols Preparation Method: in vitro wheat germ expression system Purification: Glutathione Sepharose 4 Fast Flow Storage Buffer: 50 mM Tris-HCI, 10 mM reduced Glutathione, pH=8.0 in the elution buffer. Storage Instruction: Store at -80°C. Aliquot to avoid repeated freezing and thawing. Entrez GeneID: 5606 Gene Symbol: MAP2K3 Gene Alias: MAPKK3, MEK3, MKK3, PRKMK3 Gene Summary: The protein encoded by this gene is a dual specificity protein kinase that belongs to the MAP kinase kinase family. This kinase is activated by mitogenic and environmental stress, and participates in the MAP kinase-mediated signaling cascade. -

Application of a MYC Degradation
SCIENCE SIGNALING | RESEARCH ARTICLE CANCER Copyright © 2019 The Authors, some rights reserved; Application of a MYC degradation screen identifies exclusive licensee American Association sensitivity to CDK9 inhibitors in KRAS-mutant for the Advancement of Science. No claim pancreatic cancer to original U.S. Devon R. Blake1, Angelina V. Vaseva2, Richard G. Hodge2, McKenzie P. Kline3, Thomas S. K. Gilbert1,4, Government Works Vikas Tyagi5, Daowei Huang5, Gabrielle C. Whiten5, Jacob E. Larson5, Xiaodong Wang2,5, Kenneth H. Pearce5, Laura E. Herring1,4, Lee M. Graves1,2,4, Stephen V. Frye2,5, Michael J. Emanuele1,2, Adrienne D. Cox1,2,6, Channing J. Der1,2* Stabilization of the MYC oncoprotein by KRAS signaling critically promotes the growth of pancreatic ductal adeno- carcinoma (PDAC). Thus, understanding how MYC protein stability is regulated may lead to effective therapies. Here, we used a previously developed, flow cytometry–based assay that screened a library of >800 protein kinase inhibitors and identified compounds that promoted either the stability or degradation of MYC in a KRAS-mutant PDAC cell line. We validated compounds that stabilized or destabilized MYC and then focused on one compound, Downloaded from UNC10112785, that induced the substantial loss of MYC protein in both two-dimensional (2D) and 3D cell cultures. We determined that this compound is a potent CDK9 inhibitor with a previously uncharacterized scaffold, caused MYC loss through both transcriptional and posttranslational mechanisms, and suppresses PDAC anchorage- dependent and anchorage-independent growth. We discovered that CDK9 enhanced MYC protein stability 62 through a previously unknown, KRAS-independent mechanism involving direct phosphorylation of MYC at Ser . -

Integrated Molecular Characterisation of the MAPK Pathways in Human
ARTICLE https://doi.org/10.1038/s42003-020-01552-6 OPEN Integrated molecular characterisation of the MAPK pathways in human cancers reveals pharmacologically vulnerable mutations and gene dependencies 1234567890():,; ✉ Musalula Sinkala 1 , Panji Nkhoma 2, Nicola Mulder 1 & Darren Patrick Martin1 The mitogen-activated protein kinase (MAPK) pathways are crucial regulators of the cellular processes that fuel the malignant transformation of normal cells. The molecular aberrations which lead to cancer involve mutations in, and transcription variations of, various MAPK pathway genes. Here, we examine the genome sequences of 40,848 patient-derived tumours representing 101 distinct human cancers to identify cancer-associated mutations in MAPK signalling pathway genes. We show that patients with tumours that have mutations within genes of the ERK-1/2 pathway, the p38 pathways, or multiple MAPK pathway modules, tend to have worse disease outcomes than patients with tumours that have no mutations within the MAPK pathways genes. Furthermore, by integrating information extracted from various large-scale molecular datasets, we expose the relationship between the fitness of cancer cells after CRISPR mediated gene knockout of MAPK pathway genes, and their dose-responses to MAPK pathway inhibitors. Besides providing new insights into MAPK pathways, we unearth vulnerabilities in specific pathway genes that are reflected in the re sponses of cancer cells to MAPK targeting drugs: a revelation with great potential for guiding the development of innovative therapies. -

Temperature Regulates NF-Κb Dynamics and Function Through Timing of A20 Transcription
Temperature regulates NF-κB dynamics and function through timing of A20 transcription C. V. Harpera,1, D. J. Woodcockb,c,1, C. Lama, M. Garcia-Albornozd, A. Adamsona, L. Ashalla, W. Rowea, P. Downtona, L. Schmidta, S. Weste, D. G. Spillera, D. A. Randb,c,2, and M. R. H. Whitea,2 aSystems Microscopy Centre, Division of Molecular and Cellular Function, School of Biology, Faculty of Biology, Medicine and Health, Manchester Academic Health Sciences Centre, University of Manchester, M13 9PT Manchester, United Kingdom; bMathematics Institute, University of Warwick, CV4 7AL Coventry, United Kingdom; cZeeman Institute for Systems Biology and Infectious Epidemiology Research, University of Warwick, CV4 7AL Coventry, United Kingdom; dDivision of Evolution and Genomic Sciences, School of Biology, Faculty of Biology, Medicine and Health, Manchester Academic Health Sciences Centre, University of Manchester, M13 9PT Manchester, United Kingdom; and eInstitute of Integrative Biology, University of Liverpool, L69 7ZB United Kingdom Edited by Ronald N. Germain, National Institutes of Health, Bethesda, MD, and approved April 27, 2018 (received for review March 7, 2018) NF-κB signaling plays a pivotal role in control of the inflammatory by measuring both the oscillatory dynamics and the downstream gene response. We investigated how the dynamics and function of NF-κB expression response. were affected by temperature within the mammalian physiological range (34 °C to 40 °C). An increase in temperature led to an increase Results in NF-κB nuclear/cytoplasmic oscillation frequency following Tumor To investigate the effect of temperature on NF-κB dynamics, we Necrosis Factor alpha (TNFα) stimulation. Mathematical modeling used time-lapse live-cell microscopy to study cytoplasmic to nu- suggested that this temperature sensitivity might be due to an clear oscillations of NF-κB across the physiological temper- A20-dependent mechanism, and A20 silencing removed the sensi- ature range, 34 °C to 40 °C. -
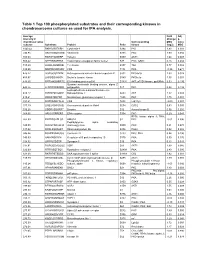
Table 1 Top 100 Phosphorylated Substrates and Their Corresponding Kinases in Chondrosarcoma Cultures As Used for IPA Analysis
Table 1 Top 100 phosphorylated substrates and their corresponding kinases in chondrosarcoma cultures as used for IPA analysis. Average Fold Adj intensity in Change p- chondrosarcoma Corresponding MSC value cultures Substrate Protein Psite kinase (log2) MSC 1043.42 RKKKVSSTKRH Cytohesin-1 S394 PKC 1.83 0.001 746.95 RKGYRSQRGHS Vitronectin S381 PKC 1.00 0.056 709.03 RARSTSLNERP Tuberin S939 AKT1 1.64 0.008 559.42 SPPRSSLRRSS Transcription elongation factor A-like1 S37 PKC; GSK3 0.18 0.684 515.29 LRRSLSRSMSQ Telethonin S157 Titin 0.77 0.082 510.00 MQPDNSSDSDY CD5 T434 PKA -0.35 0.671 476.27 GGRGGSRARNL Heterogeneous nuclear ribonucleoprotein K S302 PKCdelta 1.03 0.028 455.97 LKPGSSHRKTK Bruton's tyrosine kinase S180 PKCbeta 1.55 0.001 444.65 RRRMASMQRTG E1A binding protein p300 S1834 AKT; p70S6 kinase; pp90Rsk 0.53 0.195 Guanine nucleotide binding protein, alpha Z 440.26 HLRSESQRQRR polypeptide S27 PKC 0.88 0.199 6-phosphofructo-2-kinase/fructose-2,6- 424.12 RPRNYSVGSRP biphosphatase 2 S483 AKT 1.32 0.003 419.61 KKKIATRKPRF Metabotropic glutamate receptor 1 T695 PKC 1.75 0.001 391.21 DNSSDSDYDLH CD5 T453 Lck; Fyn -2.09 0.001 377.39 LRQLRSPRRAQ Ras associated protein Rab4 S204 CDC2 0.63 0.091 376.28 SSQRVSSYRRT Desmin S12 Aurora kinase B 0.56 0.255 369.05 ARIGGSRRERS EP4 receptor S354 PKC 0.29 0.543 RPS6 kinase alpha 3; PKA; 367.99 EPKRRSARLSA HMG14 S7 PKC -0.01 0.996 Peptidylglycine alpha amidating 349.08 SRKGYSRKGFD monooxygenase S930 PKC 0.21 0.678 347.92 RRRLSSLRAST Ribosomal protein S6 S236 PAK2 0.02 0.985 346.84 RSNPPSRKGSG Connexin -

Knockdown of Mitogen-Activated Protein Kinase Kinase 3 Negatively Regulates Hepatitis a Virus Replication
International Journal of Molecular Sciences Article Knockdown of Mitogen-Activated Protein Kinase Kinase 3 Negatively Regulates Hepatitis A Virus Replication Tatsuo Kanda 1,* , Reina Sasaki-Tanaka 1, Ryota Masuzaki 1 , Naoki Matsumoto 1, Hiroaki Okamoto 2 and Mitsuhiko Moriyama 1 1 Division of Gastroenterology and Hepatology, Department of Medicine, Nihon University School of Medicine, 30-1 Oyaguchi-kamicho, Itabashi-ku, Tokyo 173-8610, Japan; [email protected] (R.S.-T.); [email protected] (R.M.); [email protected] (N.M.); [email protected] (M.M.) 2 Division of Virology, Department of Infection and Immunity, Jichi Medical University School of Medicine, Shimotsuke, Tochigi 329-0498, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-3-3972-8111 Abstract: Zinc chloride is known to be effective in combatting hepatitis A virus (HAV) infection, and zinc ions seem to be especially involved in Toll-like receptor (TLR) signaling pathways. In the present study, we examined this involvement in human hepatoma cell lines using a human TLR signaling target RT-PCR array. We also observed that zinc chloride inhibited mitogen-activated protein kinase kinase 3 (MAP2K3) expression, which could downregulate HAV replication in human hepatocytes. It is possible that zinc chloride may inhibit HAV replication in association with its inhibition of MAP2K3. In that regard, this study set out to determine whether MAP2K3 could be considered a modulating factor in the development of the HAV pathogen-associated molecular pattern (PAMP) Citation: Kanda, T.; Sasaki-Tanaka, and its triggering of interferon-β production. -

A Novel Glycogen Synthase Kinase-3 Inhibitor Optimized for Acute
Published OnlineFirst May 9, 2016; DOI: 10.1158/1535-7163.MCT-15-0566 Small Molecule Therapeutics Molecular Cancer Therapeutics A Novel Glycogen Synthase Kinase-3 Inhibitor Optimized for Acute Myeloid Leukemia Differentiation Activity Sophia Hu1, Masumi Ueda2, Lindsay Stetson1, James Ignatz-Hoover1, Stephen Moreton1, Amit Chakrabarti3, Zhiqiang Xia3, Goutam Karan3, Marcos de Lima2, Mukesh K. Agrawal3,4, and David N. Wald1,3,5 Abstract Standard therapies used for the treatment of acute myeloid describe the discovery of a novel GSK3 inhibitor, GS87. GS87 leukemia (AML) are cytotoxic agents that target rapidly prolifer- was discovered in efforts to optimize GSK3 inhibition for AML ating cells. Unfortunately, this therapeutic approach has differentiation activity. Despite GS87's dramatic ability to induce limited efficacy and significant toxicity and the majority of AML AML differentiation, kinase profiling reveals its high specificity patients still die of their disease. In contrast to the poor prognosis in targeting GSK3 as compared with other kinases. GS87 demon- of most AML patients, most individuals with a rare subtype of strates high efficacy in a mouse AML model system and unlike AML, acute promyelocytic leukemia, can be cured by differenti- current AML therapeutics, exhibits little effect on normal bone ation therapy using regimens containing all-trans retinoic acid. marrow cells. GS87 induces potent differentiation by more GSK3 has been previously identified as a therapeutic target in effectively activating GSK3-dependent signaling components AML where its inhibition can lead to the differentiation and including MAPK signaling as compared with other GSK3 growth arrest of leukemic cells. Unfortunately, existing GSK3 inhibitors. GS87 is a novel GSK3 inhibitor with therapeutic inhibitors lead to suboptimal differentiation activity making potential as a differentiation agent for non-promyelocytic AML. -

Genome-Wide RNA Interference Screening Reveals a COPI-MAP2K3 Pathway Required for YAP Regulation
Genome-wide RNA interference screening reveals a COPI-MAP2K3 pathway required for YAP regulation Yong Joon Kima,b,1, Eunji Junga,1, Eunbie Shina, Sin-Hyoung Hongc,d, Hui Su Jeonge, Gayeong Hura,f, Hye Yun Jeonga, Seung-Hyo Leea, Ji Eun Leee,g,2, Gun-Hwa Kimc,d,2, and Joon Kima,2 aGraduate School of Medical Science and Engineering, Korea Advanced Institute of Science and Technology, 34141 Daejeon, Korea; bDepartment of Ophthalmology, Institute of Vision Research, Severance Hospital, Yonsei University College of Medicine, 06273 Seoul, Korea; cDrug & Disease Target Team, Division of Bioconvergence Analysis, Korea Basic Science Institute, 28119 Cheongju, Korea; dDepartment of Bio-Analytical Science, University of Science and Technology, 34113 Daejeon, Korea; eDepartment of Health Sciences and Technology, Samsung Advanced Institute for Health Sciences & Technology, Sungkyunkwan University, 06355 Seoul, Korea; fR&D Division, GenoFocus Inc., 34014 Daejeon, Korea; and gSamsung Biomedical Research Institute, Samsung Medical Center, 06351 Seoul, Korea Edited by Akira Suzuki, Medical Institute of Bioregulation, Kyushu University, Fukuoka, Japan, and accepted by Editorial Board Member Tak W. Mak July9, 2020 (received for review September 4, 2019) The transcriptional regulator YAP, which plays important roles in Currently, our understanding of the Hippo-independent regulation of the development, regeneration, and tumorigenesis, is activated YAP/TAZ is incomplete in part because there is no comprehensive when released from inhibition by the Hippo kinase cascade. The list of genes involved in the regulation. regulatory mechanism of YAP in Hippo-low contexts is poorly un- Here, we systematically investigate Hippo-independent YAP/ derstood. Here, we performed a genome-wide RNA interference TAZ regulation using genome-wide RNA interference (RNAi) screen to identify genes whose loss of function in a Hippo-null phenotypic screens. -

First Insight Into the Modulation of Noncanonical NF-Κb Signaling
pathogens Article First Insight into the Modulation of Noncanonical NF-κB Signaling Components by Poxviruses in Established Immune-Derived Cell Lines: An In Vitro Model of Ectromelia Virus Infection Justyna Struzik 1,*, Lidia Szulc-D ˛abrowska 1, Matylda B. Mielcarska 1, Magdalena Bossowska-Nowicka 1, Michał Koper 2 and Małgorzata Giery ´nska 1 1 Division of Immunology, Department of Preclinical Sciences, Institute of Veterinary Medicine, Warsaw University of Life Sciences-SGGW, Ciszewskiego 8, 02-786 Warsaw, Poland; [email protected] (L.S.-D.); [email protected] (M.B.M.); [email protected] (M.B.-N.); [email protected] (M.G.) 2 Institute of Genetics and Biotechnology, Faculty of Biology, University of Warsaw, A. Pawi´nskiego5A, 02-106 Warsaw, Poland; [email protected] * Correspondence: [email protected]; Tel.: +48-22-59-360-61 Received: 17 August 2020; Accepted: 1 October 2020; Published: 4 October 2020 Abstract: Dendritic cells (DCs) and macrophages are the first line of antiviral immunity. Viral pathogens exploit these cell populations for their efficient replication and dissemination via the modulation of intracellular signaling pathways. Disruption of the noncanonical nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) signaling has frequently been observed in lymphoid cells upon infection with oncogenic viruses. However, several nononcogenic viruses have been shown to manipulate the noncanonical NF-κB signaling in different cell types. This study demonstrates the modulating effect of ectromelia virus (ECTV) on the components of the noncanonical NF-κB signaling pathway in established murine cell lines: JAWS II DCs and RAW 264.7 macrophages. -

Computer Vision Profiling of Neurite Outgrowth Dynamics Reveals Spatiotemporal Modularity of Rho Gtpase Signaling
Published January 4, 2016 JCB: ArticleTools Computer vision profiling of neurite outgrowth dynamics reveals spatiotemporal modularity of Rho GTPase signaling Ludovico Fusco,1* Riwal Lefort,2* Kevin Smith,3* Fethallah Benmansour,3 German Gonzalez,3 Caterina Barillari,4 Bernd Rinn,4 Francois Fleuret,2 Pascal Fua,3 and Olivier Pertz1 1Department of Biomedicine, University of Basel, 4058 Basel, Switzerland 2Institut Dalla Molle d'Intelligence Artificielle Perceptive (IDI AP Research Institute), 1920 Martigny, Switzerland 3Computer Vision Laboratory, École Polytechnique Fédérale de Lausanne, 1015 Lausanne, Switzerland 4Department of Biosystems Science and Engineering, Eidgenössische Technische Hochschule, 4058 Basel, Switzerland Rho guanosine triphosphatases (GTPases) control the cytoskeletal dynamics that power neurite outgrowth. This process consists of dynamic neurite initiation, elongation, retraction, and branching cycles that are likely to be regulated by Downloaded from specific spatiotemporal signaling networks, which cannot be resolved with static, steady-state assays. We present Neu- riteTracker, a computer-vision approach to automatically segment and track neuronal morphodynamics in time-lapse datasets. Feature extraction then quantifies dynamic neurite outgrowth phenotypes. We identify a set of stereotypic neurite outgrowth morphodynamic behaviors in a cultured neuronal cell system. Systematic RNA interference perturba- tion of a Rho GTPase interactome consisting of 219 proteins reveals a limited set of morphodynamic phenotypes. As proof of concept, we show that loss of function of two distinct RhoA-specific GTPase-activating proteins (GAPs) leads to jcb.rupress.org opposite neurite outgrowth phenotypes. Imaging of RhoA activation dynamics indicates that both GAPs regulate differ- ent spatiotemporal Rho GTPase pools, with distinct functions. Our results provide a starting point to dissect spatiotempo- ral Rho GTPase signaling networks that regulate neurite outgrowth. -

Oxidized Phospholipids Regulate Amino Acid Metabolism Through MTHFD2 to Facilitate Nucleotide Release in Endothelial Cells
ARTICLE DOI: 10.1038/s41467-018-04602-0 OPEN Oxidized phospholipids regulate amino acid metabolism through MTHFD2 to facilitate nucleotide release in endothelial cells Juliane Hitzel1,2, Eunjee Lee3,4, Yi Zhang 3,5,Sofia Iris Bibli2,6, Xiaogang Li7, Sven Zukunft 2,6, Beatrice Pflüger1,2, Jiong Hu2,6, Christoph Schürmann1,2, Andrea Estefania Vasconez1,2, James A. Oo1,2, Adelheid Kratzer8,9, Sandeep Kumar 10, Flávia Rezende1,2, Ivana Josipovic1,2, Dominique Thomas11, Hector Giral8,9, Yannick Schreiber12, Gerd Geisslinger11,12, Christian Fork1,2, Xia Yang13, Fragiska Sigala14, Casey E. Romanoski15, Jens Kroll7, Hanjoong Jo 10, Ulf Landmesser8,9,16, Aldons J. Lusis17, 1234567890():,; Dmitry Namgaladze18, Ingrid Fleming2,6, Matthias S. Leisegang1,2, Jun Zhu 3,4 & Ralf P. Brandes1,2 Oxidized phospholipids (oxPAPC) induce endothelial dysfunction and atherosclerosis. Here we show that oxPAPC induce a gene network regulating serine-glycine metabolism with the mitochondrial methylenetetrahydrofolate dehydrogenase/cyclohydrolase (MTHFD2) as a cau- sal regulator using integrative network modeling and Bayesian network analysis in human aortic endothelial cells. The cluster is activated in human plaque material and by atherogenic lipo- proteins isolated from plasma of patients with coronary artery disease (CAD). Single nucleotide polymorphisms (SNPs) within the MTHFD2-controlled cluster associate with CAD. The MTHFD2-controlled cluster redirects metabolism to glycine synthesis to replenish purine nucleotides. Since endothelial cells secrete purines in response to oxPAPC, the MTHFD2- controlled response maintains endothelial ATP. Accordingly, MTHFD2-dependent glycine synthesis is a prerequisite for angiogenesis. Thus, we propose that endothelial cells undergo MTHFD2-mediated reprogramming toward serine-glycine and mitochondrial one-carbon metabolism to compensate for the loss of ATP in response to oxPAPC during atherosclerosis. -

Network Pharmacology Modeling Identifies Synergistic Aurora B And
www.nature.com/npjsba ARTICLE OPEN Network pharmacology modeling identifies synergistic Aurora B and ZAK interaction in triple-negative breast cancer Jing Tang 1,2,3, Prson Gautam 1, Abhishekh Gupta1,4, Liye He1, Sanna Timonen1, Yevhen Akimov1, Wenyu Wang1, Agnieszka Szwajda1, Alok Jaiswal1, Denes Turei5, Bhagwan Yadav1,6, Matti Kankainen1,7, Jani Saarela1, Julio Saez-Rodriguez5,8, Krister Wennerberg1,9 and Tero Aittokallio 1,3 Cancer cells with heterogeneous mutation landscapes and extensive functional redundancy easily develop resistance to monotherapies by emerging activation of compensating or bypassing pathways. To achieve more effective and sustained clinical responses, synergistic interactions of multiple druggable targets that inhibit redundant cancer survival pathways are often required. Here, we report a systematic polypharmacology strategy to predict, test, and understand the selective drug combinations for MDA- MB-231 triple-negative breast cancer cells. We started by applying our network pharmacology model to predict synergistic drug combinations. Next, by utilizing kinome-wide drug-target profiles and gene expression data, we pinpointed a synergistic target interaction between Aurora B and ZAK kinase inhibition that led to enhanced growth inhibition and cytotoxicity, as validated by combinatorial siRNA, CRISPR/Cas9, and drug combination experiments. The mechanism of such a context-specific target interaction was elucidated using a dynamic simulation of MDA-MB-231 signaling network, suggesting a cross-talk between p53 and p38