Regulated ADAM17-Dependent EGF Family Ligand Release by Substrate-Selecting Signaling Pathways
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Neprilysin Is Required for Angiotensin-(1-7)
Page 1 of 39 Diabetes NEPRILYSIN IS REQUIRED FOR ANGIOTENSIN-(1-7)’S ABILITY TO ENHANCE INSULIN SECRETION VIA ITS PROTEOLYTIC ACTIVITY TO GENERATE ANGIOTENSIN-(1-2) Gurkirat S. Brara, Breanne M. Barrowa, Matthew Watsonb, Ryan Griesbachc, Edwina Chounga, Andrew Welchc, Bela Ruzsicskad, Daniel P. Raleighb, Sakeneh Zraikaa,c aVeterans Affairs Puget Sound Health Care System, Seattle, WA 98108, United States bDepartment of Chemistry, Stony Brook University, Stony Brook, NY 11794, United States cDivision of Metabolism, Endocrinology and Nutrition, Department of Medicine, University of Washington, Seattle, WA 98195, United States dInstitute for Chemical Biology and Drug Discovery, Stony Brook University, Stony Brook, NY 11794, United States Short Title: Angiotensin-(1-7) and insulin secretion Word count: 3997; Figure count: 8 main (plus 3 Online Suppl.); Table count: 1 Online Suppl. Correspondence to: Sakeneh Zraika, PhD 1660 South Columbian Way (151) Seattle, WA, United States Tel: 206-768-5391 / Fax: 206-764-2164 Email: [email protected] 1 Diabetes Publish Ahead of Print, published online May 30, 2017 Diabetes Page 2 of 39 ABSTRACT Recent work has renewed interest in therapies targeting the renin-angiotensin system (RAS) to improve β-cell function in type 2 diabetes. Studies show that generation of angiotensin-(1-7) by angiotensin converting enzyme 2 (ACE2) and its binding to the Mas receptor (MasR) improves glucose homeostasis, partly by enhancing glucose-stimulated insulin secretion (GSIS). Thus, islet ACE2 upregulation is viewed as a desirable therapeutic goal. Here, we show that although endogenous islet ACE2 expression is sparse, its inhibition abrogates angiotensin-(1-7)-mediated GSIS. However, a more widely expressed islet peptidase, neprilysin, degrades angiotensin-(1-7) into several peptides. -

Cell-Autonomous FLT3L Shedding Via ADAM10 Mediates Conventional Dendritic Cell Development in Mouse Spleen
Cell-autonomous FLT3L shedding via ADAM10 mediates conventional dendritic cell development in mouse spleen Kohei Fujitaa,b,1, Svetoslav Chakarovc,1, Tetsuro Kobayashid, Keiko Sakamotod, Benjamin Voisind, Kaibo Duanc, Taneaki Nakagawaa, Keisuke Horiuchie, Masayuki Amagaib, Florent Ginhouxc, and Keisuke Nagaod,2 aDepartment of Dentistry and Oral Surgery, Keio University School of Medicine, Tokyo 160-8582, Japan; bDepartment of Dermatology, Keio University School of Medicine, Tokyo 160-8582, Japan; cSingapore Immunology Network, Agency for Science, Technology and Research, Biopolis, 138648 Singapore; dDermatology Branch, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, MD 20892; and eDepartment of Orthopedic Surgery, National Defense Medical College, Tokorozawa 359-8513, Japan Edited by Kenneth M. Murphy, Washington University School of Medicine, St. Louis, MO, and approved June 10, 2019 (received for review November 4, 2018) Conventional dendritic cells (cDCs) derive from bone marrow (BM) intocDC1sorcDC2stakesplaceintheBM(3),andthese precursors that undergo cascades of developmental programs to pre-cDC1s and pre-cDC2s ultimately differentiate into cDC1s terminally differentiate in peripheral tissues. Pre-cDC1s and pre- and cDC2s after migrating to nonlymphoid and lymphoid tissues. + + cDC2s commit in the BM to each differentiate into CD8α /CD103 cDCs are short-lived, and their homeostatic maintenance relies + cDC1s and CD11b cDC2s, respectively. Although both cDCs rely on on constant replenishment from the BM precursors (5). The cy- the cytokine FLT3L during development, mechanisms that ensure tokine Fms-related tyrosine kinase 3 ligand (FLT3L) (12), by cDC accessibility to FLT3L have yet to be elucidated. Here, we gen- signaling through its receptor FLT3 expressed on DC precursors, erated mice that lacked a disintegrin and metalloproteinase (ADAM) is essential during the development of DCs (7, 13). -
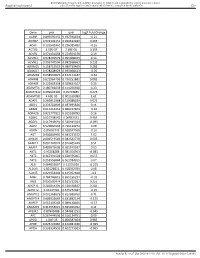
Gene Pval Qval Log2 Fold Change AAMP 0.895690332 0.952598834
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut Gene pval qval Log2 Fold Change AAMP 0.895690332 0.952598834 -0.21 ABI3BP 0.002302151 0.020612283 0.465 ACHE 0.103542461 0.296385483 -0.16 ACTG2 2.99E-07 7.68E-05 3.195 ACVR1 0.071431098 0.224504378 0.19 ACVR1C 0.978209579 0.995008423 0.14 ACVRL1 0.006747504 0.042938663 0.235 ADAM15 0.158715519 0.380719469 0.285 ADAM17 0.978208929 0.995008423 -0.05 ADAM28 0.038932876 0.152174187 -0.62 ADAM8 0.622964796 0.790251882 0.085 ADAM9 0.122003358 0.329623107 0.25 ADAMTS1 0.180766659 0.414256926 0.23 ADAMTS12 0.009902195 0.05703885 0.425 ADAMTS8 4.60E-05 0.001169089 1.61 ADAP1 0.269811968 0.519388039 0.075 ADD1 0.233702809 0.487695826 0.11 ADM2 0.012213453 0.066227879 -0.36 ADRA2B 0.822777921 0.915518785 0.16 AEBP1 0.010738542 0.06035531 0.465 AGGF1 0.117946691 0.320915024 -0.095 AGR2 0.529860903 0.736120272 0.08 AGRN 0.85693743 0.928047568 -0.16 AGT 0.006849995 0.043233572 1.02 AHNAK 0.006519543 0.042542779 0.605 AKAP12 0.001747074 0.016405449 0.51 AKAP2 0.409929603 0.665919397 0.05 AKT1 0.95208288 0.985354963 -0.085 AKT2 0.367391504 0.620376005 0.055 AKT3 0.253556844 0.501934205 0.07 ALB 0.064833867 0.21195036 -0.315 ALDOA 0.83128831 0.918352939 0.08 ALOX5 0.029954404 0.125352668 -0.3 AMH 0.784746815 0.895196237 -0.03 ANG 0.050500474 0.181732067 0.255 ANGPT1 0.281853305 0.538528647 0.285 ANGPT2 0.43147281 0.675272487 -0.15 ANGPTL2 0.001368876 0.013688762 0.71 ANGPTL4 0.686032669 0.831882134 -0.175 ANPEP 0.019103243 0.089148466 -0.57 ANXA2P2 0.412553021 0.665966092 0.11 AP1M2 0.87843088 0.944681253 -0.045 APC 0.267444505 0.516134751 0.09 APOD 1.04E-05 0.000587404 0.985 APOE 0.023722987 0.104981036 -0.395 APOH 0.336334555 0.602273505 -0.065 Sundar R, et al. -

Gene Standard Deviation MTOR 0.12553731 PRPF38A
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut Gene Standard Deviation MTOR 0.12553731 PRPF38A 0.141472605 EIF2B4 0.154700091 DDX50 0.156333027 SMC3 0.161420017 NFAT5 0.166316903 MAP2K1 0.166585267 KDM1A 0.16904912 RPS6KB1 0.170330192 FCF1 0.170391706 MAP3K7 0.170660513 EIF4E2 0.171572093 TCEB1 0.175363093 CNOT10 0.178975095 SMAD1 0.179164705 NAA15 0.179904998 SETD2 0.180182498 HDAC3 0.183971158 AMMECR1L 0.184195031 CHD4 0.186678211 SF3A3 0.186697697 CNOT4 0.189434633 MTMR14 0.189734199 SMAD4 0.192451524 TLK2 0.192702667 DLG1 0.19336621 COG7 0.193422331 SP1 0.194364189 PPP3R1 0.196430217 ERBB2IP 0.201473001 RAF1 0.206887192 CUL1 0.207514271 VEZF1 0.207579584 SMAD3 0.208159809 TFDP1 0.208834504 VAV2 0.210269344 ADAM17 0.210687138 SMURF2 0.211437666 MRPS5 0.212428684 TMUB2 0.212560675 SRPK2 0.216217428 MAP2K4 0.216345366 VHL 0.219735582 SMURF1 0.221242495 PLCG1 0.221688351 EP300 0.221792349 Sundar R, et al. Gut 2020;0:1–10. doi: 10.1136/gutjnl-2020-320805 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut MGAT5 0.222050228 CDC42 0.2230598 DICER1 0.225358787 RBX1 0.228272533 ZFYVE16 0.22831803 PTEN 0.228595789 PDCD10 0.228799406 NF2 0.23091035 TP53 0.232683696 RB1 0.232729172 TCF20 0.2346075 PPP2CB 0.235117302 AGK 0.235416298 -

CD Markers Are Routinely Used for the Immunophenotyping of Cells
ptglab.com 1 CD MARKER ANTIBODIES www.ptglab.com Introduction The cluster of differentiation (abbreviated as CD) is a protocol used for the identification and investigation of cell surface molecules. So-called CD markers are routinely used for the immunophenotyping of cells. Despite this use, they are not limited to roles in the immune system and perform a variety of roles in cell differentiation, adhesion, migration, blood clotting, gamete fertilization, amino acid transport and apoptosis, among many others. As such, Proteintech’s mini catalog featuring its antibodies targeting CD markers is applicable to a wide range of research disciplines. PRODUCT FOCUS PECAM1 Platelet endothelial cell adhesion of blood vessels – making up a large portion molecule-1 (PECAM1), also known as cluster of its intracellular junctions. PECAM-1 is also CD Number of differentiation 31 (CD31), is a member of present on the surface of hematopoietic the immunoglobulin gene superfamily of cell cells and immune cells including platelets, CD31 adhesion molecules. It is highly expressed monocytes, neutrophils, natural killer cells, on the surface of the endothelium – the thin megakaryocytes and some types of T-cell. Catalog Number layer of endothelial cells lining the interior 11256-1-AP Type Rabbit Polyclonal Applications ELISA, FC, IF, IHC, IP, WB 16 Publications Immunohistochemical of paraffin-embedded Figure 1: Immunofluorescence staining human hepatocirrhosis using PECAM1, CD31 of PECAM1 (11256-1-AP), Alexa 488 goat antibody (11265-1-AP) at a dilution of 1:50 anti-rabbit (green), and smooth muscle KD/KO Validated (40x objective). alpha-actin (red), courtesy of Nicola Smart. PECAM1: Customer Testimonial Nicola Smart, a cardiovascular researcher “As you can see [the immunostaining] is and a group leader at the University of extremely clean and specific [and] displays Oxford, has said of the PECAM1 antibody strong intercellular junction expression, (11265-1-AP) that it “worked beautifully as expected for a cell adhesion molecule.” on every occasion I’ve tried it.” Proteintech thanks Dr. -

The Disintegrin/Metalloproteinase ADAM10 Is Essential for the Establishment of the Brain Cortex
The Journal of Neuroscience, April 7, 2010 • 30(14):4833–4844 • 4833 Development/Plasticity/Repair The Disintegrin/Metalloproteinase ADAM10 Is Essential for the Establishment of the Brain Cortex Ellen Jorissen,1,2* Johannes Prox,3* Christian Bernreuther,4 Silvio Weber,3 Ralf Schwanbeck,3 Lutgarde Serneels,1,2 An Snellinx,1,2 Katleen Craessaerts,1,2 Amantha Thathiah,1,2 Ina Tesseur,1,2 Udo Bartsch,5 Gisela Weskamp,6 Carl P. Blobel,6 Markus Glatzel,4 Bart De Strooper,1,2 and Paul Saftig3 1Center for Human Genetics, Katholieke Universiteit Leuven and 2Department for Developmental and Molecular Genetics, Vlaams Instituut voor Biotechnologie (VIB), 3000 Leuven, Belgium, 3Institut fu¨r Biochemie, Christian-Albrechts-Universita¨t zu Kiel, D-24098 Kiel, Germany, 4Institute of Neuropathology, University Medical Center Hamburg Eppendorf, 20246 Hamburg, Germany, 5Department of Ophthalmology, University Medical Center Hamburg Eppendorf, 20246 Hamburg, Germany, and 6Arthritis and Tissue Degeneration Program, Hospital for Special Surgery, and Departments of Medicine and of Physiology, Systems Biology and Biophysics, Weill Medical College of Cornell University, New York, New York 10021 The metalloproteinase and major amyloid precursor protein (APP) ␣-secretase candidate ADAM10 is responsible for the shedding of ,proteins important for brain development, such as cadherins, ephrins, and Notch receptors. Adam10 ؊/؊ mice die at embryonic day 9.5 due to major defects in development of somites and vasculogenesis. To investigate the function of ADAM10 in brain, we generated Adam10conditionalknock-out(cKO)miceusingaNestin-Crepromotor,limitingADAM10inactivationtoneuralprogenitorcells(NPCs) and NPC-derived neurons and glial cells. The cKO mice die perinatally with a disrupted neocortex and a severely reduced ganglionic eminence, due to precocious neuronal differentiation resulting in an early depletion of progenitor cells. -

Cleavage of the Extracellular Domain of Junctional Adhesion Molecule-A Is Associated with Resistance to Anti-HER2 Therapies in Breast Cancer Settings Astrid O
Leech et al. Breast Cancer Research (2018) 20:140 https://doi.org/10.1186/s13058-018-1064-1 RESEARCH ARTICLE Open Access Cleavage of the extracellular domain of junctional adhesion molecule-A is associated with resistance to anti-HER2 therapies in breast cancer settings Astrid O. Leech1, Sri HariKrishna Vellanki1†, Emily J. Rutherford1†, Aoife Keogh1, Hanne Jahns2, Lance Hudson1, Norma O’Donovan3, Siham Sabri4, Bassam Abdulkarim5, Katherine M. Sheehan6, Elaine W. Kay6, Leonie S. Young1, Arnold D. K. Hill1, Yvonne E. Smith1 and Ann M. Hopkins1* Abstract Background: Junctional adhesion molecule-A (JAM-A) is an adhesion molecule whose overexpression on breast tumor tissue has been associated with aggressive cancer phenotypes, including human epidermal growth factor receptor-2 (HER2)-positive disease. Since JAM-A has been described to regulate HER2 expression in breast cancer cells, we hypothesized that JAM-dependent stabilization of HER2 could participate in resistance to HER2-targeted therapies. Methods: Using breast cancer cell line models resistant to anti-HER2 drugs, we investigated JAM-A expression and the effect of JAM-A silencing on biochemical/functional parameters. We also tested whether altered JAM-A expression/ processing underpinned differences between drug-sensitive and -resistant cells and acted as a biomarker of patients who developed resistance to HER2-targeted therapies. Results: Silencing JAM-A enhanced the anti-proliferative effects of anti-HER2 treatments in trastuzumab- and lapatinib- resistant breast cancer cells and further reduced HER2 protein expression and Akt phosphorylation in drug-treated cells. Increased epidermal growth factor receptor expression observed in drug-resistant models was normalized upon JAM-A silencing. -

Strategies to Target ADAM17 in Disease: from Its Discovery to the Irhom Revolution
molecules Review Strategies to Target ADAM17 in Disease: From Its Discovery to the iRhom Revolution Matteo Calligaris 1,2,†, Doretta Cuffaro 2,†, Simone Bonelli 1, Donatella Pia Spanò 3, Armando Rossello 2, Elisa Nuti 2,* and Simone Dario Scilabra 1,* 1 Proteomics Group of Fondazione Ri.MED, Research Department IRCCS ISMETT (Istituto Mediterraneo per i Trapianti e Terapie ad Alta Specializzazione), Via E. Tricomi 5, 90145 Palermo, Italy; [email protected] (M.C.); [email protected] (S.B.) 2 Department of Pharmacy, University of Pisa, Via Bonanno 6, 56126 Pisa, Italy; [email protected] (D.C.); [email protected] (A.R.) 3 Università degli Studi di Palermo, STEBICEF (Dipartimento di Scienze e Tecnologie Biologiche Chimiche e Farmaceutiche), Viale delle Scienze Ed. 16, 90128 Palermo, Italy; [email protected] * Correspondence: [email protected] (E.N.); [email protected] (S.D.S.) † These authors contributed equally to this work. Abstract: For decades, disintegrin and metalloproteinase 17 (ADAM17) has been the object of deep investigation. Since its discovery as the tumor necrosis factor convertase, it has been considered a major drug target, especially in the context of inflammatory diseases and cancer. Nevertheless, the development of drugs targeting ADAM17 has been harder than expected. This has generally been due to its multifunctionality, with over 80 different transmembrane proteins other than tumor necrosis factor α (TNF) being released by ADAM17, and its structural similarity to other metalloproteinases. This review provides an overview of the different roles of ADAM17 in disease and the effects of its ablation in a number of in vivo models of pathological conditions. -

Proteolytic Cleavage—Mechanisms, Function
Review Cite This: Chem. Rev. 2018, 118, 1137−1168 pubs.acs.org/CR Proteolytic CleavageMechanisms, Function, and “Omic” Approaches for a Near-Ubiquitous Posttranslational Modification Theo Klein,†,⊥ Ulrich Eckhard,†,§ Antoine Dufour,†,¶ Nestor Solis,† and Christopher M. Overall*,†,‡ † ‡ Life Sciences Institute, Department of Oral Biological and Medical Sciences, and Department of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada ABSTRACT: Proteases enzymatically hydrolyze peptide bonds in substrate proteins, resulting in a widespread, irreversible posttranslational modification of the protein’s structure and biological function. Often regarded as a mere degradative mechanism in destruction of proteins or turnover in maintaining physiological homeostasis, recent research in the field of degradomics has led to the recognition of two main yet unexpected concepts. First, that targeted, limited proteolytic cleavage events by a wide repertoire of proteases are pivotal regulators of most, if not all, physiological and pathological processes. Second, an unexpected in vivo abundance of stable cleaved proteins revealed pervasive, functionally relevant protein processing in normal and diseased tissuefrom 40 to 70% of proteins also occur in vivo as distinct stable proteoforms with undocumented N- or C- termini, meaning these proteoforms are stable functional cleavage products, most with unknown functional implications. In this Review, we discuss the structural biology aspects and mechanisms -

Regulation of Profibrotic Responses by ADAM17 Activation in High Glucose Requires Its C-Terminus and FAK Renzhong Li, Tony Wang, Khyati Walia, Bo Gao and Joan C
© 2018. Published by The Company of Biologists Ltd | Journal of Cell Science (2018) 131, jcs208629. doi:10.1242/jcs.208629 RESEARCH ARTICLE Regulation of profibrotic responses by ADAM17 activation in high glucose requires its C-terminus and FAK Renzhong Li, Tony Wang, Khyati Walia, Bo Gao and Joan C. Krepinsky* ABSTRACT and amphiregulin have been implicated in the pathogenesis of Glomerular matrix accumulation is the hallmark of diabetic diabetic kidney disease (Kefaloyianni et al., 2016; Melenhorst et al., nephropathy. The metalloprotease ADAM17 mediates high glucose 2009; Uttarwar et al., 2011). Cleavage of the renoprotective (HG)-induced matrix production by kidney mesangial cells through exopeptidase ACE2 by ADAM17 is also thought to contribute to release of ligands for the epidermal growth factor receptor. Here, diabetic kidney tubular injury (Salem et al., 2014). In diabetic mice, we study the mechanism by which HG activates ADAM17. We find a nonselective metalloprotease inhibitor decreased glomerular that the C-terminus is essential for ADAM17 activation and the sclerosis, although effects cannot be specifically attributed to profibrotic response to HG. In the C-terminus, Src-mediated Y702 inhibition of ADAM17 (Ford et al., 2013). More specific ADAM17 phosphorylation and PI3K–MEK–Erk-mediated T735 phosphorylation inhibitors, however, have been associated with adverse effects, are crucial for ADAM17 activation, both are also required for the preventing their long-term use in the treatment of diabetic HG-induced increase in cell surface -

Xo PANEL DNA GENE LIST
xO PANEL DNA GENE LIST ~1700 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes (at 400 -500x average coverage on tumor) Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 -

Multi-Antigen Imaging Reveals Inflammatory DC, ADAM17
International Journal of Molecular Sciences Article Multi-Antigen Imaging Reveals Inflammatory DC, ADAM17 and Neprilysin as Effectors in Keloid Formation Mathias Rath 1 , Alain Pitiot 2, Michael Kirr 3,4,5, Waltraud Fröhlich 3,4,5, Bianca Plosnita 6, Stefan Schliep 3,4,5, Jürgen Bauerschmitz 3,4,5, Andreas S. Baur 3,4,5 and Christian Ostalecki 3,4,5,7,* 1 Department of Urology, University Hospital Heidelberg, 69120 Heidelberg, Germany; [email protected] 2 Laboratory of Image & Data Analysis, Ilixa Ltd., London W1U 6NQ, UK; [email protected] 3 Department of Dermatology, Universitätsklinikum Erlangen, Friedrich-Alexander-University Erlangen-Nürnberg, 91054 Erlangen, Germany; [email protected] (M.K.); [email protected] (W.F.); [email protected] (S.S.); [email protected] (J.B.); [email protected] (A.S.B.) 4 Deutsches Zentrum Immuntherapie (DZI), 91054 Erlangen, Germany 5 Comprehensive Cancer Center (CCC) Erlangen, 91054 Erlangen, Germany 6 TissueGnostics GmbH, 1020 Vienna, Austria; [email protected] 7 Department of Psychiatry and Psychotherapy, Universitätsklinikum Erlangen, Friedrich-Alexander-University Erlangen-Nürnberg, 91054 Erlangen, Germany * Correspondence: [email protected]; Tel.: +49-9131-8532965 Abstract: Keloid is an aberrant scarring process of the skin, characterized by excessive extracellular matrix synthesis and deposition. The pathogenesis of this prevalent cutaneous disorder is not fully understood; however, a persistent inflammatory process is observed. To obtain more insight into Citation: Rath, M.; Pitiot, A.; Kirr, M.; this process, we analyzed lesional, perilesional and healthy tissue using multi-antigen-analysis Fröhlich, W.; Plosnita, B.; Schliep, S.; (MAA) in conjunction with a data mining approach.