Neprilysin Is Required for Angiotensin-(1-7)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

View a Copy of This Licence, Visit
Choi et al. BMC Cardiovascular Disorders (2020) 20:360 https://doi.org/10.1186/s12872-020-01636-5 RESEARCH ARTICLE Open Access Soluble neprilysin and long-term clinical outcomes in patients with coronary artery disease undergoing percutaneous coronary intervention: a retrospective cohort study Ik Jun Choi1, Sungmin Lim2* , Youngdeok Hwang3, Dongjae Lee1, Won Jik Lee1, Kwan Yong Lee1, Mi-Jeong Kim1 and Doo Soo Jeon1 Abstract Background: Neprilysin has an essential role in regulating fluid balance and vascular resistance, and neprilysin inhibitors have shown beneficial effects in patients with heart failure. However, the potential predictive value of neprilysin levels as a biomarker for cardiovascular risk remains unclear. The aim of this study was to assess the prognostic value of soluble neprilysin (sNEP) levels in patients with ischemic heart disease. Methods: Neprilysin levels were measured in 694 consecutive patients with coronary artery disease (CAD) undergoing percutaneous coronary intervention (PCI). These patients were classified into two groups according to their serum levels of neprilysin and categorized into the lower neprilysin group (n = 348) and the higher neprilysin group (n = 346). The primary clinical endpoint was all-cause mortality, and the secondary endpoint was a composite of major adverse cardiac events (MACE). Results: The median sNEP level was 76.0 pg/ml. The median sNEP levels were higher in patients with left ventricular ejection fraction (LVEF) ≥40% (77.6 pg/ml, interquartile range 46.6–141.3) than in those with LVEF < 40% (70.0 pg/ml, interquartile range 47.1–100.6; P = 0.032). Among all patients, each clinical outcome and MACE did not differ significantly according to the groups divided into median, tertile, or quartile of sNEP levels during a median follow-up of 28.4 months. -

Stromal CD10 Expression in Breast Cancer Correlates with Tumor Invasion and Cancer Stem Cell Phenotype
Louhichi et al. BMC Cancer (2018) 18:49 DOI 10.1186/s12885-017-3951-8 RESEARCH ARTICLE Open Access Stromal CD10 expression in breast cancer correlates with tumor invasion and cancer stem cell phenotype Tahani Louhichi, Hanene Saad, Myriam Ben Dhiab, Sonia Ziadi and Mounir Trimeche* Abstract Background: Previous investigations have indicated that CD10 is associated with biological aggressivity in human cancers, but the use of this marker for diagnosis and prognosis is more complex. The aim of this study was to evaluate the expression of CD10 in breast cancer and its association with the clinicopathological features. In addition, we investigated whether a relationship exists between CD10 expression and cancer stem cells. Methods: CD10 expression was examined by the immunohistochemistry in a series of 133 invasive breast carcinoma cases. Results were correlated to several clinicopathological parameters. Cancer stem cell phenotype was assessed by the immunohistochemical analysis of CD44 and ALDH1. Results: Significant CD10 expression was found in the fusiform stromal cells in 19.5% of the cases and in the neoplastic cells in 7% of the cases. The stromal CD10 positivity was more frequently found in tumors with lymph node metastasis (p = 0.01) and a high histological grade (p = 0.01). However, CD10 expression by the neoplastic cells correlates with a high histological grade (p = 0.03) and the absence of estrogen (p = 0.002) as well as progesterone (p = 0.001) receptor expression. We also found that CD10 expression by the stromal cells, but not by the neoplastic cells, correlates significantly with the expression of cancer stem cell markers (CD44+/ALDH1+) (p = 0.002). -

Angiotensin Receptor Neprilysin Inhibition (ARNI) Following Acute Myocardial Infarction: Primary Results of the PARADISE-MI Trial Marc A
Angiotensin Receptor Neprilysin Inhibition (ARNI) Following Acute Myocardial Infarction: Primary Results of the PARADISE-MI Trial Marc A. Pfeffer, MD, PhD Distinguished Dzau Professor of Medicine Harvard Medical School Cardiovascular Division, Brigham and Women’s Hospital for the PARADISE-MI Committees, National Leaders and Investigators SAVE AIRE TRACE Radionuclide Clinical and/or Echocardiographic EF ≤ 40% radiographic signs EF ≤ 35% (1992) of HF (1993) (1995) 0.4 All-Cause Mortality 0.35 0.3 0.25 Placebo ACE-I 0.2 0.15 Placebo: 866/2971 (29.1%) Probability of of Probability Event 0.1 ACE-I: 702/2995 (23.4%) 0.05 OR: 0.74 (0.66–0.83) 0 Years 0 1 2 3 4 ACE-I 2995 2250 1617 892 223 Placebo 2971 2184 1521 853 138 Flather MD, et al. Lancet. 2000;355:1575–1581 Mortality in SAVE, TRACE, AIRE, and VALIANT Favors Active Drug Pfeffer,Pfeffer, McMurray, McMurray, Velazquez, Velazquez, et etal. al. N NEngl Engl J MedJ Med2003;3492003;349 2014 40 Enalapril 1117 32 (n=4212) 914 24 LCZ696 (n=4187) Meier Estimate of Meier Estimate 16 - HR = 0.80 (0.73-0.87) Cumulative (%) Rates Cumulative 8 Kaplan P = 0.0000002 Number needed to treat = 21 0 0 180 360 540 720 900 1080 1260 Patients at Risk Days After Randomization LCZ696 4187 3922 3663 3018 2257 1544 896 249 Enalapril 4212 3883 3579 2922 2123 1488 853 236 McMurray, N Engl J Med. 2014 AMI (0.5-7 days with LVEF ≤40% and/or pulmonary congestion) PLUS any risk enhancer Age ≥70 years Atrial fibrillation eGFR <60 LVEF < 30% Diabetes Killip class ≥III Prior MI STEMI without reperfusion Major Exclusions: Prior HF Clinical instability eGFR <30 Sacubitril/Valsartan Ramipril No run-in Target 97/103 mg BID Target 5 mg BID double-blind -controlled N=2830 active N=2831 Event driven: 711 primary endpoints Median follow-up: 23 months Primary Endpoint: CV death, HF hospitalization, outpatient development of HF Jering, Eur J ACC.21 Secondary Endpoint: CV death or first HF hospitalization Heart Fail. -

Cell-Autonomous FLT3L Shedding Via ADAM10 Mediates Conventional Dendritic Cell Development in Mouse Spleen
Cell-autonomous FLT3L shedding via ADAM10 mediates conventional dendritic cell development in mouse spleen Kohei Fujitaa,b,1, Svetoslav Chakarovc,1, Tetsuro Kobayashid, Keiko Sakamotod, Benjamin Voisind, Kaibo Duanc, Taneaki Nakagawaa, Keisuke Horiuchie, Masayuki Amagaib, Florent Ginhouxc, and Keisuke Nagaod,2 aDepartment of Dentistry and Oral Surgery, Keio University School of Medicine, Tokyo 160-8582, Japan; bDepartment of Dermatology, Keio University School of Medicine, Tokyo 160-8582, Japan; cSingapore Immunology Network, Agency for Science, Technology and Research, Biopolis, 138648 Singapore; dDermatology Branch, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, MD 20892; and eDepartment of Orthopedic Surgery, National Defense Medical College, Tokorozawa 359-8513, Japan Edited by Kenneth M. Murphy, Washington University School of Medicine, St. Louis, MO, and approved June 10, 2019 (received for review November 4, 2018) Conventional dendritic cells (cDCs) derive from bone marrow (BM) intocDC1sorcDC2stakesplaceintheBM(3),andthese precursors that undergo cascades of developmental programs to pre-cDC1s and pre-cDC2s ultimately differentiate into cDC1s terminally differentiate in peripheral tissues. Pre-cDC1s and pre- and cDC2s after migrating to nonlymphoid and lymphoid tissues. + + cDC2s commit in the BM to each differentiate into CD8α /CD103 cDCs are short-lived, and their homeostatic maintenance relies + cDC1s and CD11b cDC2s, respectively. Although both cDCs rely on on constant replenishment from the BM precursors (5). The cy- the cytokine FLT3L during development, mechanisms that ensure tokine Fms-related tyrosine kinase 3 ligand (FLT3L) (12), by cDC accessibility to FLT3L have yet to be elucidated. Here, we gen- signaling through its receptor FLT3 expressed on DC precursors, erated mice that lacked a disintegrin and metalloproteinase (ADAM) is essential during the development of DCs (7, 13). -
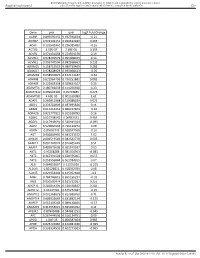
Gene Pval Qval Log2 Fold Change AAMP 0.895690332 0.952598834
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut Gene pval qval Log2 Fold Change AAMP 0.895690332 0.952598834 -0.21 ABI3BP 0.002302151 0.020612283 0.465 ACHE 0.103542461 0.296385483 -0.16 ACTG2 2.99E-07 7.68E-05 3.195 ACVR1 0.071431098 0.224504378 0.19 ACVR1C 0.978209579 0.995008423 0.14 ACVRL1 0.006747504 0.042938663 0.235 ADAM15 0.158715519 0.380719469 0.285 ADAM17 0.978208929 0.995008423 -0.05 ADAM28 0.038932876 0.152174187 -0.62 ADAM8 0.622964796 0.790251882 0.085 ADAM9 0.122003358 0.329623107 0.25 ADAMTS1 0.180766659 0.414256926 0.23 ADAMTS12 0.009902195 0.05703885 0.425 ADAMTS8 4.60E-05 0.001169089 1.61 ADAP1 0.269811968 0.519388039 0.075 ADD1 0.233702809 0.487695826 0.11 ADM2 0.012213453 0.066227879 -0.36 ADRA2B 0.822777921 0.915518785 0.16 AEBP1 0.010738542 0.06035531 0.465 AGGF1 0.117946691 0.320915024 -0.095 AGR2 0.529860903 0.736120272 0.08 AGRN 0.85693743 0.928047568 -0.16 AGT 0.006849995 0.043233572 1.02 AHNAK 0.006519543 0.042542779 0.605 AKAP12 0.001747074 0.016405449 0.51 AKAP2 0.409929603 0.665919397 0.05 AKT1 0.95208288 0.985354963 -0.085 AKT2 0.367391504 0.620376005 0.055 AKT3 0.253556844 0.501934205 0.07 ALB 0.064833867 0.21195036 -0.315 ALDOA 0.83128831 0.918352939 0.08 ALOX5 0.029954404 0.125352668 -0.3 AMH 0.784746815 0.895196237 -0.03 ANG 0.050500474 0.181732067 0.255 ANGPT1 0.281853305 0.538528647 0.285 ANGPT2 0.43147281 0.675272487 -0.15 ANGPTL2 0.001368876 0.013688762 0.71 ANGPTL4 0.686032669 0.831882134 -0.175 ANPEP 0.019103243 0.089148466 -0.57 ANXA2P2 0.412553021 0.665966092 0.11 AP1M2 0.87843088 0.944681253 -0.045 APC 0.267444505 0.516134751 0.09 APOD 1.04E-05 0.000587404 0.985 APOE 0.023722987 0.104981036 -0.395 APOH 0.336334555 0.602273505 -0.065 Sundar R, et al. -

Epigenetic Suppression of Neprilysin Regulates Breast Cancer Invasion
OPEN Citation: Oncogenesis (2016) 5, e207; doi:10.1038/oncsis.2016.16 www.nature.com/oncsis ORIGINAL ARTICLE Epigenetic suppression of neprilysin regulates breast cancer invasion HM Stephen, RJ Khoury, PR Majmudar, T Blaylock1, K Hawkins1, MS Salama1, MD Scott1, B Cosminsky1, NK Utreja1, J Britt and RE Conway In women, invasive breast cancer is the second most common cancer and the second cause of cancer-related death. Therefore, identifying novel regulators of breast cancer invasion could lead to additional biomarkers and therapeutic targets. Neprilysin, a cell-surface enzyme that cleaves and inactivates a number of substrates including endothelin-1 (ET1), has been implicated in breast cancer, but whether neprilysin promotes or inhibits breast cancer cell progression and metastasis is unclear. Here, we asked whether neprilysin expression predicts and functionally regulates breast cancer cell invasion. RT–PCR and flow cytometry analysis of MDA-MB-231 and MCF-7 breast cancer cell lines revealed decreased neprilysin expression compared with normal epithelial cells. Expression was also suppressed in invasive ductal carcinoma (IDC) compared with normal tissue. In addition, in vitro invasion assays demonstrated that neprilysin overexpression decreased breast cancer cell invasion, whereas neprilysin suppression augmented invasion. Furthermore, inhibiting neprilysin in MCF-7 breast cancer cells increased ET1 levels significantly, whereas overexpressing neprilysin decreased extracellular-signal related kinase (ERK) activation, indicating that neprilysin negatively regulates ET1-induced activation of mitogen-activated protein kinase (MAPK) signaling. To determine whether neprilysin was epigenetically suppressed in breast cancer, we performed bisulfite conversion analysis of breast cancer cells and clinical tumor samples. We found that the neprilysin promoter was hypermethylated in breast cancer; chemical reversal of methylation in MDA-MB-231 cells reactivated neprilysin expression and inhibited cancer cell invasion. -

R&D Assay for Alzheimer's Disease
R&DR&D assayassay forfor Alzheimer’sAlzheimer’s diseasedisease Target screening⳼ Ⲽ㬔 antibody array, ᢜ⭉㬔 ⸽ἐⴐ Amyloid β-peptide Alzheimer’s disease⯸ ኸᷠ᧔ ᆹ⸽ inhibitor, antibody, ELISA kit Surwhrph#Surilohu#Dqwlerg|#Duud| 6OUSFBUFE 1."5SFBUFE )41 $3&# &3, &3, )41 $3&# &3, &3, 壤伡庰䋸TBNQMF ɅH 侴䋸嵄䍴䋸BOBMZUFT䋸䬱娴哜塵 1$ 1$ 1$ 1$ 5IFNPTUSFGFSFODFEBSSBZT 1$ 1$ QQ α 34, .4, 503 Q α 34, .4, 503 %SVHTDSFFOJOH0òUBSHFUFòFDUT0ATHWAY涭廐 6OUSFBUFE 堄币䋸4BNQMF侴䋸8FTUFSOPS&-*4"䍘䧽 1."5SFBUFE P 8FTUFSOCMPU廽喜儤应侴䋸0, Z 4VCTUSBUF -JHIU )31DPOKVHBUFE1BO "OUJQIPTQIPUZSPTJOF .FBO1JYFM%FOTJUZ Y $BQUVSF"OUJCPEZ 5BSHFU"OBMZUF "SSBZ.FNCSBOF $3&# &3, &3, )41 .4, Q α 34, 503 Human XL Cytokine Array kit (ARY022, 102 analytes) Adiponectin,Aggrecan,Angiogenin,Angiopoietin-1,Angiopoietin-2,BAFF,BDNF,Complement,Component C5/C5a,CD14,CD30,CD40L, Chitinase 3-like 1,Complement Factor D,C-Reactive Protein,Cripto-1,Cystatin C,Dkk-1,DPPIV,EGF,EMMPRIN,ENA-78,Endoglin, Fas L,FGF basic,FGF- 7,FGF-19,Flt-3 L,G-CSF,GDF-15,GM-CSF,GRO-α,Grow th Hormone,HGF,ICAM-1,IFN-γ,IGFBP-2,IGFBP-3, IL-1α,IL-1β, IL-1ra,IL-2,IL-3,IL-4,IL- 5,IL-6,IL-8, IL-10,IL-11,IL-12, IL-13,IL-15,IL-16,IL-17A,IL-18 BPa,IL-19,IL-22, IL-23,IL-24,IL-27, IL-31,IL-32α/β/γ,IL-33,IL-34,IP-10,I-TAC,Kallikrein 3,Leptin,LIF,Lipocalin-2,MCP-1,MCP-3,M-CSF,MIF,MIG,MIP-1α/MIP-1β,MIP-3α,MIP-3β,MMP-9, Myeloperoxidase,Osteopontin, p70, PDGF-AA, PDGF-AB/BB,Pentraxin-3, PF4, RAGE, RANTES,RBP4,Relaxin-2, Resistin,SDF-1α,Serpin E1, SHBG, ST2, TARC,TFF3,TfR,TGF- ,Thrombospondin-1,TNF-α, uPAR, VEGF, Vitamin D BP Human Protease (34 analytes) / -

Gene Standard Deviation MTOR 0.12553731 PRPF38A
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut Gene Standard Deviation MTOR 0.12553731 PRPF38A 0.141472605 EIF2B4 0.154700091 DDX50 0.156333027 SMC3 0.161420017 NFAT5 0.166316903 MAP2K1 0.166585267 KDM1A 0.16904912 RPS6KB1 0.170330192 FCF1 0.170391706 MAP3K7 0.170660513 EIF4E2 0.171572093 TCEB1 0.175363093 CNOT10 0.178975095 SMAD1 0.179164705 NAA15 0.179904998 SETD2 0.180182498 HDAC3 0.183971158 AMMECR1L 0.184195031 CHD4 0.186678211 SF3A3 0.186697697 CNOT4 0.189434633 MTMR14 0.189734199 SMAD4 0.192451524 TLK2 0.192702667 DLG1 0.19336621 COG7 0.193422331 SP1 0.194364189 PPP3R1 0.196430217 ERBB2IP 0.201473001 RAF1 0.206887192 CUL1 0.207514271 VEZF1 0.207579584 SMAD3 0.208159809 TFDP1 0.208834504 VAV2 0.210269344 ADAM17 0.210687138 SMURF2 0.211437666 MRPS5 0.212428684 TMUB2 0.212560675 SRPK2 0.216217428 MAP2K4 0.216345366 VHL 0.219735582 SMURF1 0.221242495 PLCG1 0.221688351 EP300 0.221792349 Sundar R, et al. Gut 2020;0:1–10. doi: 10.1136/gutjnl-2020-320805 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut MGAT5 0.222050228 CDC42 0.2230598 DICER1 0.225358787 RBX1 0.228272533 ZFYVE16 0.22831803 PTEN 0.228595789 PDCD10 0.228799406 NF2 0.23091035 TP53 0.232683696 RB1 0.232729172 TCF20 0.2346075 PPP2CB 0.235117302 AGK 0.235416298 -

CD Markers Are Routinely Used for the Immunophenotyping of Cells
ptglab.com 1 CD MARKER ANTIBODIES www.ptglab.com Introduction The cluster of differentiation (abbreviated as CD) is a protocol used for the identification and investigation of cell surface molecules. So-called CD markers are routinely used for the immunophenotyping of cells. Despite this use, they are not limited to roles in the immune system and perform a variety of roles in cell differentiation, adhesion, migration, blood clotting, gamete fertilization, amino acid transport and apoptosis, among many others. As such, Proteintech’s mini catalog featuring its antibodies targeting CD markers is applicable to a wide range of research disciplines. PRODUCT FOCUS PECAM1 Platelet endothelial cell adhesion of blood vessels – making up a large portion molecule-1 (PECAM1), also known as cluster of its intracellular junctions. PECAM-1 is also CD Number of differentiation 31 (CD31), is a member of present on the surface of hematopoietic the immunoglobulin gene superfamily of cell cells and immune cells including platelets, CD31 adhesion molecules. It is highly expressed monocytes, neutrophils, natural killer cells, on the surface of the endothelium – the thin megakaryocytes and some types of T-cell. Catalog Number layer of endothelial cells lining the interior 11256-1-AP Type Rabbit Polyclonal Applications ELISA, FC, IF, IHC, IP, WB 16 Publications Immunohistochemical of paraffin-embedded Figure 1: Immunofluorescence staining human hepatocirrhosis using PECAM1, CD31 of PECAM1 (11256-1-AP), Alexa 488 goat antibody (11265-1-AP) at a dilution of 1:50 anti-rabbit (green), and smooth muscle KD/KO Validated (40x objective). alpha-actin (red), courtesy of Nicola Smart. PECAM1: Customer Testimonial Nicola Smart, a cardiovascular researcher “As you can see [the immunostaining] is and a group leader at the University of extremely clean and specific [and] displays Oxford, has said of the PECAM1 antibody strong intercellular junction expression, (11265-1-AP) that it “worked beautifully as expected for a cell adhesion molecule.” on every occasion I’ve tried it.” Proteintech thanks Dr. -

Characterization of Aminopeptidase in the Free-Living Nematode Panagrellus Redivivus: Subcellular Distribution and Possible Role in Neuropeptide Metabolism E
Journal of Nematology 39(2):153–160. 2007. © The Society of Nematologists 2007. Characterization of Aminopeptidase in the Free-living Nematode Panagrellus redivivus: Subcellular Distribution and Possible Role in Neuropeptide Metabolism E. P. Masler Abstract: Aminopeptidase was detected in homogenates of the free-living nematode Panagrellus redivivus with the aminoacyl substrate L-alanine-4-nitroanilide. Subcellular distribution of activity was 80% soluble and 20% membrane-associated. Aminopep- tidases in the two fractions differed in affinity for Ala-4-NA, with Km’s of 0.65 mM (soluble) and 2.90 mM (membrane). Specific activities (units/mg) at pH 7.8, 27°C were 9.10 (soluble) and 14.30 (membrane). Each enzyme was competitively inhibited by amastatin (90% at 100 µM inhibitor, IC50 = 3.7 µM) and inhibited by puromycin (30% at 500 µM) and 1,10-phenanthroline (IC50’s:; 148 µM, soluble; 89 µM, membrane). Activity was restored by Zn++, with maximum recoveries of 50% (soluble) and 90% (mem- ∼ brane), each at 23 µM ZnCl2. Estimated molecular masses for each were 150 kDa. FMRFamide-like neuropeptides behaved as competitive inhibitors. Modification of the N-terminal F of FMRFamide weakened inhibition by 95%, suggesting that the N-terminus is essential for binding to the enzyme. Two nematode FMRFamides, APKPFIRFa and RNKFEFIRFa, were the most potent tested. This is the first biochemical characterization of aminopeptidase in a free-living nematode other than Caenorhabditis elegans and demon- strates the high selectivity of the P. redivivus enzymes for neuropeptide substrates. Key words: FMRFamide-like peptide, inhibitor; membrane, metallopeptidase, neuropeptide, protease Nematodes, like other eukaryotic organisms, depend reproduction (Day and Maule, 1999; Maule et al., 2002; upon proteolytic enzymes for the regulation of essential Rogers et al., 2003). -

Changes to the Drug Formularies
Published September 2, 2021 The formularies and pharmaceutical management procedures are updated on a bimonthly basis, and the following changes reflect the decisions made in April 2021 by our Pharmacy and Therapeutics Committee. These updates are effective on the dates noted throughout this document. Please reference the guide below to navigate this communication: Section I. Highmark Commercial and Healthcare Reform Formularies A. Changes to the Highmark Comprehensive Formulary and the Highmark Comprehensive Healthcare Reform Formulary B. Changes to the Highmark Progressive Healthcare Reform Formulary C. Changes to the Highmark Healthcare Reform Essential Formulary D. Changes to the Highmark Core Formulary E. Changes to the Highmark National Select Formulary F. Updates to the Pharmacy Utilization Management Programs 1. Prior Authorization Program 2. Managed Prescription Drug Coverage (MRxC) Program 3. Formulary Program 4. Quantity Level Limit (QLL) Programs Section II. Highmark Medicare Part D Formularies A. Changes to the Highmark Medicare Part D 5-Tier Incentive Formulary B. Changes to the Highmark Medicare Part D 5-Tier Closed Formulary C. Additions to the Specialty Tier D. Updates to the Pharmacy Utilization Management Programs 1. Prior Authorization Program 2. Managed Prescription Drug Coverage (MRxC) Program 3. Quantity Level Limit (QLL) Program As an added convenience, you can also search our drug formularies and view utilization management policies on the Provider Resource Center (accessible via NaviNet® or our website). Click the Pharmacy Program/Formularies link from the menu on the left. Highmark Blue Shield is an independent licensee of the Blue Cross and Blue Shield Association. NaviNet is a registered trademark of NaviNet, Inc., which is an independent company that provides a secure, web-based portal between providers and health insurance companies. -

Mtor Signaling in Pulmonary Vascular Disease: Pathogenic Role and Therapeutic Target
International Journal of Molecular Sciences Review mTOR Signaling in Pulmonary Vascular Disease: Pathogenic Role and Therapeutic Target Aleksandra Babicheva 1, Ayako Makino 2 and Jason X.-J. Yuan 1,* 1 Section of Physiology, Division of Pulmonary, Critical Care and Sleep Medicine, Department of Medicine, University of California, San Diego, CA 92093, USA; [email protected] 2 Division of Endocrinology and Metabolism, Department of Medicine, University of California, San Diego, 9500 Gilman Drive, MC 0856, La Jolla, CA 92093-0856, USA; [email protected] * Correspondence: [email protected] Abstract: Pulmonary arterial hypertension (PAH) is a progressive and fatal disease without a cure. The exact pathogenic mechanisms of PAH are complex and poorly understood, yet a number of abnormally expressed genes and regulatory pathways contribute to sustained vasoconstriction and vascular remodeling of the distal pulmonary arteries. Mammalian target of rapamycin (mTOR) is one of the major signaling pathways implicated in regulating cell proliferation, migration, differentiation, and protein synthesis. Here we will describe the canonical mTOR pathway, structural and functional differences between mTOR complexes 1 and 2, as well as the crosstalk with other important signaling cascades in the development of PAH. The pathogenic role of mTOR in pulmonary vascular remodel- ing and sustained vasoconstriction due to its contribution to proliferation, migration, phenotypic transition, and gene regulation in pulmonary artery smooth muscle and endothelial cells will be discussed. Despite the progress in our elucidation of the etiology and pathogenesis of PAH over the two last decades, there is a lack of effective therapeutic agents to treat PAH patients representing Citation: Babicheva, A.; Makino, A.; a significant unmet clinical need.