The Role of GDF5 in the Developing Vertebrate Nervous System
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Neprilysin Is Required for Angiotensin-(1-7)
Page 1 of 39 Diabetes NEPRILYSIN IS REQUIRED FOR ANGIOTENSIN-(1-7)’S ABILITY TO ENHANCE INSULIN SECRETION VIA ITS PROTEOLYTIC ACTIVITY TO GENERATE ANGIOTENSIN-(1-2) Gurkirat S. Brara, Breanne M. Barrowa, Matthew Watsonb, Ryan Griesbachc, Edwina Chounga, Andrew Welchc, Bela Ruzsicskad, Daniel P. Raleighb, Sakeneh Zraikaa,c aVeterans Affairs Puget Sound Health Care System, Seattle, WA 98108, United States bDepartment of Chemistry, Stony Brook University, Stony Brook, NY 11794, United States cDivision of Metabolism, Endocrinology and Nutrition, Department of Medicine, University of Washington, Seattle, WA 98195, United States dInstitute for Chemical Biology and Drug Discovery, Stony Brook University, Stony Brook, NY 11794, United States Short Title: Angiotensin-(1-7) and insulin secretion Word count: 3997; Figure count: 8 main (plus 3 Online Suppl.); Table count: 1 Online Suppl. Correspondence to: Sakeneh Zraika, PhD 1660 South Columbian Way (151) Seattle, WA, United States Tel: 206-768-5391 / Fax: 206-764-2164 Email: [email protected] 1 Diabetes Publish Ahead of Print, published online May 30, 2017 Diabetes Page 2 of 39 ABSTRACT Recent work has renewed interest in therapies targeting the renin-angiotensin system (RAS) to improve β-cell function in type 2 diabetes. Studies show that generation of angiotensin-(1-7) by angiotensin converting enzyme 2 (ACE2) and its binding to the Mas receptor (MasR) improves glucose homeostasis, partly by enhancing glucose-stimulated insulin secretion (GSIS). Thus, islet ACE2 upregulation is viewed as a desirable therapeutic goal. Here, we show that although endogenous islet ACE2 expression is sparse, its inhibition abrogates angiotensin-(1-7)-mediated GSIS. However, a more widely expressed islet peptidase, neprilysin, degrades angiotensin-(1-7) into several peptides. -

Familial Juvenile Polyposis Syndrome with a De Novo Germline Missense Variant in BMPR1A Gene: a Case Report Qing Liu, Mengling Liu, Tianshu Liu and Yiyi Yu*
Liu et al. BMC Medical Genetics (2020) 21:196 https://doi.org/10.1186/s12881-020-01135-6 CASE REPORT Open Access Familial juvenile polyposis syndrome with a de novo germline missense variant in BMPR1A gene: a case report Qing Liu, Mengling Liu, Tianshu Liu and Yiyi Yu* Abstract Background: Juvenile polyposis syndrome (JPS) is a rare autosomal dominant hereditary disorder characterized by the development of multiple distinct juvenile polyps in the gastrointestinal tract with an increased risk of colorectal cancer. Germline mutations in two genes, SMAD4 and BMPR1A, have been identified to cause JPS. Case presentation: Here, we report a germline heterozygous missense variant (c.299G > A) in exon 3 BMPR1A gene in a family with juvenile polyposis. This variant was absent from the population database, and concluded as de novo compared with the parental sequencing. Further sequencing of the proband’s children confirmed the segregation of this variant with the disease, while the variant was also predicted to have damaging effect based on online prediction tools. Therefore, this variant was classified as likely pathogenic according to the American College of Medical Genetics and Genomics (ACMG) guidelines. Conclusions: Germline genetic testing revealed a de novo germline missense variant in BMPR1A gene in a family with juvenile polyposis. Identification of the pathogenic variant facilitates the cancer risk management of at-risk family members, and endoscopic surveillance is recommended for mutation carriers. Keywords: Juvenile polyposis syndrome, BMPR1A gene, De novo germline variant, Missense variant Background two genes, SMAD4 and BMPR1A, have been identi- Juvenile polyposis syndrome (JPS) is a rare autosomal fied to cause JPS [5]. -

The Prevalence of MADH4 and BMPR1A Mutations in Juvenile Polyposis and Absence of BMPR2, BMPR1B, and ACVR1 Mutations
484 ORIGINAL ARTICLE J Med Genet: first published as 10.1136/jmg.2004.018598 on 2 July 2004. Downloaded from The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations J R Howe, M G Sayed, A F Ahmed, J Ringold, J Larsen-Haidle, A Merg, F A Mitros, C A Vaccaro, G M Petersen, F M Giardiello, S T Tinley, L A Aaltonen, H T Lynch ............................................................................................................................... J Med Genet 2004;41:484–491. doi: 10.1136/jmg.2004.018598 Background: Juvenile polyposis (JP) is an autosomal dominant syndrome predisposing to colorectal and gastric cancer. We have identified mutations in two genes causing JP, MADH4 and bone morphogenetic protein receptor 1A (BMPR1A): both are involved in bone morphogenetic protein (BMP) mediated signalling and are members of the TGF-b superfamily. This study determined the prevalence of mutations See end of article for in MADH4 and BMPR1A, as well as three other BMP/activin pathway candidate genes in a large number authors’ affiliations ....................... of JP patients. Methods: DNA was extracted from the blood of JP patients and used for PCR amplification of each exon of Correspondence to: these five genes, using primers flanking each intron–exon boundary. Mutations were determined by Dr J R Howe, Department of Surgery, 4644 JCP, comparison to wild type sequences using sequence analysis software. A total of 77 JP cases were University of Iowa College sequenced for mutations in the MADH4, BMPR1A, BMPR1B, BMPR2, and/or ACVR1 (activin A receptor) of Medicine, 200 Hawkins genes. The latter three genes were analysed when MADH4 and BMPR1A sequencing found no mutations. -

Evolutionary Origin of Bone Morphogenetic Protein 15 And
Evolutionary origin of Bone Morphogenetic Protein 15 and growth and differentiation factor 9 and differential selective pressure between mono- and polyovulating species Olivier Monestier, Bertrand Servin, Sylvain Auclair, Thomas Bourquard, Anne Poupon, Géraldine Pascal, Stéphane Fabre To cite this version: Olivier Monestier, Bertrand Servin, Sylvain Auclair, Thomas Bourquard, Anne Poupon, et al.. Evo- lutionary origin of Bone Morphogenetic Protein 15 and growth and differentiation factor 9 and differ- ential selective pressure between mono- and polyovulating species. Biology of Reproduction, Society for the Study of Reproduction, 2014, 91 (4), pp.1-13. 10.1095/biolreprod.114.119735. hal-01129871 HAL Id: hal-01129871 https://hal.archives-ouvertes.fr/hal-01129871 Submitted on 27 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. BIOLOGY OF REPRODUCTION (2014) 91(4):83, 1–13 Published online before print 6 August 2014. DOI 10.1095/biolreprod.114.119735 Evolutionary Origin of Bone Morphogenetic Protein 15 and Growth and Differentiation Factor 9 -

Cell-Autonomous FLT3L Shedding Via ADAM10 Mediates Conventional Dendritic Cell Development in Mouse Spleen
Cell-autonomous FLT3L shedding via ADAM10 mediates conventional dendritic cell development in mouse spleen Kohei Fujitaa,b,1, Svetoslav Chakarovc,1, Tetsuro Kobayashid, Keiko Sakamotod, Benjamin Voisind, Kaibo Duanc, Taneaki Nakagawaa, Keisuke Horiuchie, Masayuki Amagaib, Florent Ginhouxc, and Keisuke Nagaod,2 aDepartment of Dentistry and Oral Surgery, Keio University School of Medicine, Tokyo 160-8582, Japan; bDepartment of Dermatology, Keio University School of Medicine, Tokyo 160-8582, Japan; cSingapore Immunology Network, Agency for Science, Technology and Research, Biopolis, 138648 Singapore; dDermatology Branch, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, MD 20892; and eDepartment of Orthopedic Surgery, National Defense Medical College, Tokorozawa 359-8513, Japan Edited by Kenneth M. Murphy, Washington University School of Medicine, St. Louis, MO, and approved June 10, 2019 (received for review November 4, 2018) Conventional dendritic cells (cDCs) derive from bone marrow (BM) intocDC1sorcDC2stakesplaceintheBM(3),andthese precursors that undergo cascades of developmental programs to pre-cDC1s and pre-cDC2s ultimately differentiate into cDC1s terminally differentiate in peripheral tissues. Pre-cDC1s and pre- and cDC2s after migrating to nonlymphoid and lymphoid tissues. + + cDC2s commit in the BM to each differentiate into CD8α /CD103 cDCs are short-lived, and their homeostatic maintenance relies + cDC1s and CD11b cDC2s, respectively. Although both cDCs rely on on constant replenishment from the BM precursors (5). The cy- the cytokine FLT3L during development, mechanisms that ensure tokine Fms-related tyrosine kinase 3 ligand (FLT3L) (12), by cDC accessibility to FLT3L have yet to be elucidated. Here, we gen- signaling through its receptor FLT3 expressed on DC precursors, erated mice that lacked a disintegrin and metalloproteinase (ADAM) is essential during the development of DCs (7, 13). -

Tumor Promoting Effect of BMP Signaling in Endometrial Cancer
International Journal of Molecular Sciences Article Tumor Promoting Effect of BMP Signaling in Endometrial Cancer Tomohiko Fukuda 1,* , Risa Fukuda 1, Kohei Miyazono 1,2,† and Carl-Henrik Heldin 1,*,† 1 Science for Life Laboratory, Department of Medical Biochemistry and Microbiology, Box 582, Uppsala University, SE-751 23 Uppsala, Sweden; [email protected] (R.F.); [email protected] (K.M.) 2 Department of Molecular Pathology, Graduate School of Medicine, The University of Tokyo, Tokyo 113-0033, Japan * Correspondence: [email protected] (T.F.); [email protected] (C.-H.H.); Tel.: +46-18-4714738 (T.F.); +46-18-4714738 (C.-H.H.) † These authors contributed equally to this work. Abstract: The effects of bone morphogenetic proteins (BMPs), members of the transforming growth factor-β (TGF-β) family, in endometrial cancer (EC) have yet to be determined. In this study, we analyzed the TCGA and MSK-IMPACT datasets and investigated the effects of BMP2 and of TWSG1, a BMP antagonist, on Ishikawa EC cells. Frequent ACVR1 mutations and high mRNA expressions of BMP ligands and receptors were observed in EC patients of the TCGA and MSK-IMPACT datasets. Ishikawa cells secreted higher amounts of BMP2 compared with ovarian cancer cell lines. Exogenous BMP2 stimulation enhanced EC cell sphere formation via c-KIT induction. BMP2 also induced EMT of EC cells, and promoted migration by induction of SLUG. The BMP receptor kinase inhibitor LDN193189 augmented the growth inhibitory effects of carboplatin. Analyses of mRNAs of several BMP antagonists revealed that TWSG1 mRNA was abundantly expressed in Ishikawa cells. -

Identification of Two Novel Mutations in the NOG Gene Associated With
Journal of Human Genetics (2015) 60, 27–34 & 2015 The Japan Society of Human Genetics All rights reserved 1434-5161/15 www.nature.com/jhg ORIGINAL ARTICLE Identification of two novel mutations in the NOG gene associated with congenital stapes ankylosis and symphalangism Akira Ganaha1,3, Tadashi Kaname2,3, Yukinori Akazawa1,3, Teruyuki Higa1,3, Ayano Shinjou1,3, Kenji Naritomi2,3 and Mikio Suzuki1,3 In this study, we describe three unrelated Japanese patients with hearing loss and symphalangism who were diagnosed with proximal symphalangism (SYM1), atypical multiple synostosis syndrome (atypical SYNS1) and stapes ankylosis with broad thumb and toes (SABTT), respectively, based on the clinical features. Surgical findings in the middle ear were similar among the patients. By next-generation and Sanger sequencing analyses, we identified two novel mutations, c.559C4G (p.P178A) and c.682T4A (p.C228S), in the SYM1 and atypical SYNS1 families, respectively. No pathogenic changes were found in the protein-coding regions, exon–intron boundaries or promoter regions of the NOG, GDF5 or FGF9 genes in the SABTT family. Such negative molecular data suggest there may be further genetic heterogeneity underlying SYNS1, with the involvement of at least one additional gene. Stapedotomy resulted in good hearing in all patients over the long term, indicating no correlation between genotype and surgical outcome. Given the overlap of the clinical features of these syndromes in our patients and the molecular findings, the diagnostic term ‘NOG-related-symphalangism spectrum disorder (NOG-SSD)’ is advocated and an unidentified gene may be responsible for this disorder. Journal of Human Genetics (2015) 60, 27–34; doi:10.1038/jhg.2014.97; published online 13 November 2014 INTRODUCTION However, as the GDF5 protein interacts with the NOG protein,14,15 Proximal symphalangism (SYM1) with conductive hearing loss was the diagnostic category of NOG-SSD does appear to be promising. -

Supplemental Table 1. Complete Gene Lists and GO Terms from Figure 3C
Supplemental Table 1. Complete gene lists and GO terms from Figure 3C. Path 1 Genes: RP11-34P13.15, RP4-758J18.10, VWA1, CHD5, AZIN2, FOXO6, RP11-403I13.8, ARHGAP30, RGS4, LRRN2, RASSF5, SERTAD4, GJC2, RHOU, REEP1, FOXI3, SH3RF3, COL4A4, ZDHHC23, FGFR3, PPP2R2C, CTD-2031P19.4, RNF182, GRM4, PRR15, DGKI, CHMP4C, CALB1, SPAG1, KLF4, ENG, RET, GDF10, ADAMTS14, SPOCK2, MBL1P, ADAM8, LRP4-AS1, CARNS1, DGAT2, CRYAB, AP000783.1, OPCML, PLEKHG6, GDF3, EMP1, RASSF9, FAM101A, STON2, GREM1, ACTC1, CORO2B, FURIN, WFIKKN1, BAIAP3, TMC5, HS3ST4, ZFHX3, NLRP1, RASD1, CACNG4, EMILIN2, L3MBTL4, KLHL14, HMSD, RP11-849I19.1, SALL3, GADD45B, KANK3, CTC- 526N19.1, ZNF888, MMP9, BMP7, PIK3IP1, MCHR1, SYTL5, CAMK2N1, PINK1, ID3, PTPRU, MANEAL, MCOLN3, LRRC8C, NTNG1, KCNC4, RP11, 430C7.5, C1orf95, ID2-AS1, ID2, GDF7, KCNG3, RGPD8, PSD4, CCDC74B, BMPR2, KAT2B, LINC00693, ZNF654, FILIP1L, SH3TC1, CPEB2, NPFFR2, TRPC3, RP11-752L20.3, FAM198B, TLL1, CDH9, PDZD2, CHSY3, GALNT10, FOXQ1, ATXN1, ID4, COL11A2, CNR1, GTF2IP4, FZD1, PAX5, RP11-35N6.1, UNC5B, NKX1-2, FAM196A, EBF3, PRRG4, LRP4, SYT7, PLBD1, GRASP, ALX1, HIP1R, LPAR6, SLITRK6, C16orf89, RP11-491F9.1, MMP2, B3GNT9, NXPH3, TNRC6C-AS1, LDLRAD4, NOL4, SMAD7, HCN2, PDE4A, KANK2, SAMD1, EXOC3L2, IL11, EMILIN3, KCNB1, DOK5, EEF1A2, A4GALT, ADGRG2, ELF4, ABCD1 Term Count % PValue Genes regulation of pathway-restricted GDF3, SMAD7, GDF7, BMPR2, GDF10, GREM1, BMP7, LDLRAD4, SMAD protein phosphorylation 9 6.34 1.31E-08 ENG pathway-restricted SMAD protein GDF3, SMAD7, GDF7, BMPR2, GDF10, GREM1, BMP7, LDLRAD4, phosphorylation -
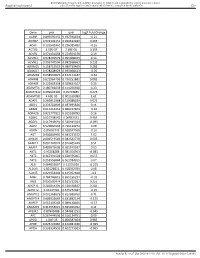
Gene Pval Qval Log2 Fold Change AAMP 0.895690332 0.952598834
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut Gene pval qval Log2 Fold Change AAMP 0.895690332 0.952598834 -0.21 ABI3BP 0.002302151 0.020612283 0.465 ACHE 0.103542461 0.296385483 -0.16 ACTG2 2.99E-07 7.68E-05 3.195 ACVR1 0.071431098 0.224504378 0.19 ACVR1C 0.978209579 0.995008423 0.14 ACVRL1 0.006747504 0.042938663 0.235 ADAM15 0.158715519 0.380719469 0.285 ADAM17 0.978208929 0.995008423 -0.05 ADAM28 0.038932876 0.152174187 -0.62 ADAM8 0.622964796 0.790251882 0.085 ADAM9 0.122003358 0.329623107 0.25 ADAMTS1 0.180766659 0.414256926 0.23 ADAMTS12 0.009902195 0.05703885 0.425 ADAMTS8 4.60E-05 0.001169089 1.61 ADAP1 0.269811968 0.519388039 0.075 ADD1 0.233702809 0.487695826 0.11 ADM2 0.012213453 0.066227879 -0.36 ADRA2B 0.822777921 0.915518785 0.16 AEBP1 0.010738542 0.06035531 0.465 AGGF1 0.117946691 0.320915024 -0.095 AGR2 0.529860903 0.736120272 0.08 AGRN 0.85693743 0.928047568 -0.16 AGT 0.006849995 0.043233572 1.02 AHNAK 0.006519543 0.042542779 0.605 AKAP12 0.001747074 0.016405449 0.51 AKAP2 0.409929603 0.665919397 0.05 AKT1 0.95208288 0.985354963 -0.085 AKT2 0.367391504 0.620376005 0.055 AKT3 0.253556844 0.501934205 0.07 ALB 0.064833867 0.21195036 -0.315 ALDOA 0.83128831 0.918352939 0.08 ALOX5 0.029954404 0.125352668 -0.3 AMH 0.784746815 0.895196237 -0.03 ANG 0.050500474 0.181732067 0.255 ANGPT1 0.281853305 0.538528647 0.285 ANGPT2 0.43147281 0.675272487 -0.15 ANGPTL2 0.001368876 0.013688762 0.71 ANGPTL4 0.686032669 0.831882134 -0.175 ANPEP 0.019103243 0.089148466 -0.57 ANXA2P2 0.412553021 0.665966092 0.11 AP1M2 0.87843088 0.944681253 -0.045 APC 0.267444505 0.516134751 0.09 APOD 1.04E-05 0.000587404 0.985 APOE 0.023722987 0.104981036 -0.395 APOH 0.336334555 0.602273505 -0.065 Sundar R, et al. -

Gene Standard Deviation MTOR 0.12553731 PRPF38A
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut Gene Standard Deviation MTOR 0.12553731 PRPF38A 0.141472605 EIF2B4 0.154700091 DDX50 0.156333027 SMC3 0.161420017 NFAT5 0.166316903 MAP2K1 0.166585267 KDM1A 0.16904912 RPS6KB1 0.170330192 FCF1 0.170391706 MAP3K7 0.170660513 EIF4E2 0.171572093 TCEB1 0.175363093 CNOT10 0.178975095 SMAD1 0.179164705 NAA15 0.179904998 SETD2 0.180182498 HDAC3 0.183971158 AMMECR1L 0.184195031 CHD4 0.186678211 SF3A3 0.186697697 CNOT4 0.189434633 MTMR14 0.189734199 SMAD4 0.192451524 TLK2 0.192702667 DLG1 0.19336621 COG7 0.193422331 SP1 0.194364189 PPP3R1 0.196430217 ERBB2IP 0.201473001 RAF1 0.206887192 CUL1 0.207514271 VEZF1 0.207579584 SMAD3 0.208159809 TFDP1 0.208834504 VAV2 0.210269344 ADAM17 0.210687138 SMURF2 0.211437666 MRPS5 0.212428684 TMUB2 0.212560675 SRPK2 0.216217428 MAP2K4 0.216345366 VHL 0.219735582 SMURF1 0.221242495 PLCG1 0.221688351 EP300 0.221792349 Sundar R, et al. Gut 2020;0:1–10. doi: 10.1136/gutjnl-2020-320805 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut MGAT5 0.222050228 CDC42 0.2230598 DICER1 0.225358787 RBX1 0.228272533 ZFYVE16 0.22831803 PTEN 0.228595789 PDCD10 0.228799406 NF2 0.23091035 TP53 0.232683696 RB1 0.232729172 TCF20 0.2346075 PPP2CB 0.235117302 AGK 0.235416298 -

CD Markers Are Routinely Used for the Immunophenotyping of Cells
ptglab.com 1 CD MARKER ANTIBODIES www.ptglab.com Introduction The cluster of differentiation (abbreviated as CD) is a protocol used for the identification and investigation of cell surface molecules. So-called CD markers are routinely used for the immunophenotyping of cells. Despite this use, they are not limited to roles in the immune system and perform a variety of roles in cell differentiation, adhesion, migration, blood clotting, gamete fertilization, amino acid transport and apoptosis, among many others. As such, Proteintech’s mini catalog featuring its antibodies targeting CD markers is applicable to a wide range of research disciplines. PRODUCT FOCUS PECAM1 Platelet endothelial cell adhesion of blood vessels – making up a large portion molecule-1 (PECAM1), also known as cluster of its intracellular junctions. PECAM-1 is also CD Number of differentiation 31 (CD31), is a member of present on the surface of hematopoietic the immunoglobulin gene superfamily of cell cells and immune cells including platelets, CD31 adhesion molecules. It is highly expressed monocytes, neutrophils, natural killer cells, on the surface of the endothelium – the thin megakaryocytes and some types of T-cell. Catalog Number layer of endothelial cells lining the interior 11256-1-AP Type Rabbit Polyclonal Applications ELISA, FC, IF, IHC, IP, WB 16 Publications Immunohistochemical of paraffin-embedded Figure 1: Immunofluorescence staining human hepatocirrhosis using PECAM1, CD31 of PECAM1 (11256-1-AP), Alexa 488 goat antibody (11265-1-AP) at a dilution of 1:50 anti-rabbit (green), and smooth muscle KD/KO Validated (40x objective). alpha-actin (red), courtesy of Nicola Smart. PECAM1: Customer Testimonial Nicola Smart, a cardiovascular researcher “As you can see [the immunostaining] is and a group leader at the University of extremely clean and specific [and] displays Oxford, has said of the PECAM1 antibody strong intercellular junction expression, (11265-1-AP) that it “worked beautifully as expected for a cell adhesion molecule.” on every occasion I’ve tried it.” Proteintech thanks Dr.