The Role of the Metalloproteases ADAM8, 9, 10 and 17 in Leukocyte Migration in Vitro
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

Neprilysin Is Required for Angiotensin-(1-7)
Page 1 of 39 Diabetes NEPRILYSIN IS REQUIRED FOR ANGIOTENSIN-(1-7)’S ABILITY TO ENHANCE INSULIN SECRETION VIA ITS PROTEOLYTIC ACTIVITY TO GENERATE ANGIOTENSIN-(1-2) Gurkirat S. Brara, Breanne M. Barrowa, Matthew Watsonb, Ryan Griesbachc, Edwina Chounga, Andrew Welchc, Bela Ruzsicskad, Daniel P. Raleighb, Sakeneh Zraikaa,c aVeterans Affairs Puget Sound Health Care System, Seattle, WA 98108, United States bDepartment of Chemistry, Stony Brook University, Stony Brook, NY 11794, United States cDivision of Metabolism, Endocrinology and Nutrition, Department of Medicine, University of Washington, Seattle, WA 98195, United States dInstitute for Chemical Biology and Drug Discovery, Stony Brook University, Stony Brook, NY 11794, United States Short Title: Angiotensin-(1-7) and insulin secretion Word count: 3997; Figure count: 8 main (plus 3 Online Suppl.); Table count: 1 Online Suppl. Correspondence to: Sakeneh Zraika, PhD 1660 South Columbian Way (151) Seattle, WA, United States Tel: 206-768-5391 / Fax: 206-764-2164 Email: [email protected] 1 Diabetes Publish Ahead of Print, published online May 30, 2017 Diabetes Page 2 of 39 ABSTRACT Recent work has renewed interest in therapies targeting the renin-angiotensin system (RAS) to improve β-cell function in type 2 diabetes. Studies show that generation of angiotensin-(1-7) by angiotensin converting enzyme 2 (ACE2) and its binding to the Mas receptor (MasR) improves glucose homeostasis, partly by enhancing glucose-stimulated insulin secretion (GSIS). Thus, islet ACE2 upregulation is viewed as a desirable therapeutic goal. Here, we show that although endogenous islet ACE2 expression is sparse, its inhibition abrogates angiotensin-(1-7)-mediated GSIS. However, a more widely expressed islet peptidase, neprilysin, degrades angiotensin-(1-7) into several peptides. -

What Are the Roles of Metalloproteinases in Cartilage and Bone Damage? G Murphy, M H Lee
iv44 Ann Rheum Dis: first published as 10.1136/ard.2005.042465 on 20 October 2005. Downloaded from REPORT What are the roles of metalloproteinases in cartilage and bone damage? G Murphy, M H Lee ............................................................................................................................... Ann Rheum Dis 2005;64:iv44–iv47. doi: 10.1136/ard.2005.042465 enzyme moiety into an upper and a lower subdomain. A A role for metalloproteinases in the pathological destruction common five stranded beta-sheet and two alpha-helices are in diseases such as rheumatoid arthritis and osteoarthritis, always found in the upper subdomain with a further C- and the irreversible nature of the ensuing cartilage and bone terminal helix in the lower subdomain. The catalytic sites of damage, have been the focus of much investigation for the metalloproteinases, especially the MMPs, have been several decades. This has led to the development of broad targeted for the development of low molecular weight spectrum metalloproteinase inhibitors as potential therapeu- synthetic inhibitors with a zinc chelating moiety. Inhibitors tics. More recently it has been appreciated that several able to fully differentiate between individual enzymes have families of zinc dependent proteinases play significant and not been identified thus far, although a reasonable level of varied roles in the biology of the resident cells in these tissues, discrimination is now being achieved in some cases.7 Each orchestrating development, remodelling, and subsequent family does, however, have other unique domains with pathological processes. They also play key roles in the numerous roles, including the determination of physiological activity of inflammatory cells. The task of elucidating the substrate specificity, ECM, or cell surface localisation (fig 1). -

Prion Fragment Peptides Are Digested with Membrane Type Matrix Metalloproteinases and Acquire Enzyme Resistance Through Cu2+-Binding
Biomolecules 2014, 4, 510-526; doi:10.3390/biom4020510 OPEN ACCESS biomolecules ISSN 2218-273X www.mdpi.com/journal/biomolecules/ Article Prion Fragment Peptides Are Digested with Membrane Type Matrix Metalloproteinases and Acquire Enzyme Resistance through Cu2+-Binding Aya Kojima 1,2, Motomi Konishi 1 and Toshifumi Akizawa 1,* 1 Analytical Chemistry, Pharmaceutical Science, Setsunan University, 45-1 Nagaotoge-cho, Hirakata, Osaka 573-0101, Japan; E-Mails: [email protected] (A.K.); [email protected] (M.K.) 2 Division of Cellular and Molecular Biology, Department of Cancer Biology, Institute of Medical Science, The University of Tokyo, 4-6-1 Shirokanedai, Minato-ku, Tokyo 180-8639, Japan * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel./Fax: +81-72-866-3129. Received: 31 January 2014; in revised form: 2 April 2014 / Accepted: 11 April 2014 / Published: 8 May 2014 Abstract: Prions are the cause of neurodegenerative disease in humans and other mammals. The structural conversion of the prion protein (PrP) from a normal cellular protein (PrPC) to a protease-resistant isoform (PrPSc) is thought to relate to Cu2+ binding to histidine residues. In this study, we focused on the membrane-type matrix metalloproteinases (MT-MMPs) such as MT1-MMP and MT3-MMP, which are expressed in the brain as PrPC-degrading proteases. We synthesized 21 prion fragment peptides. Each purified peptide was individually incubated with recombinant MT1-MMP or MT3-MMP in the presence or absence of Cu2+ and the cleavage sites determined by LC-ESI-MS analysis. Recombinant MMP-7 and human serum (HS) were also tested as control. -

Supporting Online Material
1 2 3 4 5 6 7 Supplementary Information for 8 9 Fractalkine-induced microglial vasoregulation occurs within the retina and is altered early in diabetic 10 retinopathy 11 12 *Samuel A. Mills, *Andrew I. Jobling, *Michael A. Dixon, Bang V. Bui, Kirstan A. Vessey, Joanna A. Phipps, 13 Ursula Greferath, Gene Venables, Vickie H.Y. Wong, Connie H.Y. Wong, Zheng He, Flora Hui, James C. 14 Young, Josh Tonc, Elena Ivanova, Botir T. Sagdullaev, Erica L. Fletcher 15 * Joint first authors 16 17 Corresponding author: 18 Prof. Erica L. Fletcher. Department of Anatomy & Neuroscience. The University of Melbourne, Grattan St, 19 Parkville 3010, Victoria, Australia. 20 Email: [email protected] ; Tel: +61-3-8344-3218; Fax: +61-3-9347-5219 21 22 This PDF file includes: 23 24 Supplementary text 25 Figures S1 to S10 26 Tables S1 to S7 27 Legends for Movies S1 to S2 28 SI References 29 30 Other supplementary materials for this manuscript include the following: 31 32 Movies S1 to S2 33 34 35 36 1 1 Supplementary Information Text 2 Materials and Methods 3 Microglial process movement on retinal vessels 4 Dark agouti rats were anaesthetized, injected intraperitoneally with rhodamine B (Sigma-Aldrich) to label blood 5 vessels and retinal explants established as described in the main text. Retinal microglia were labelled with Iba-1 6 and imaging performed on an inverted confocal microscope (Leica SP5). Baseline images were taken for 10 7 minutes, followed by the addition of PBS (10 minutes) and then either fractalkine or fractalkine + candesartan 8 (10 minutes) using concentrations outlined in the main text. -

3 Cleavage Products of Notch 2/Site and Myelopoiesis by Dysregulating
ADAM10 Overexpression Shifts Lympho- and Myelopoiesis by Dysregulating Site 2/Site 3 Cleavage Products of Notch This information is current as David R. Gibb, Sheinei J. Saleem, Dae-Joong Kang, Mark of October 4, 2021. A. Subler and Daniel H. Conrad J Immunol 2011; 186:4244-4252; Prepublished online 2 March 2011; doi: 10.4049/jimmunol.1003318 http://www.jimmunol.org/content/186/7/4244 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2011/03/02/jimmunol.100331 Material 8.DC1 http://www.jimmunol.org/ References This article cites 45 articles, 16 of which you can access for free at: http://www.jimmunol.org/content/186/7/4244.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on October 4, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2011 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology ADAM10 Overexpression Shifts Lympho- and Myelopoiesis by Dysregulating Site 2/Site 3 Cleavage Products of Notch David R. -

Negatively Regulated by ADAM15 TRIF-Mediated TLR3 and TLR4
TRIF-Mediated TLR3 and TLR4 Signaling Is Negatively Regulated by ADAM15 Suaad Ahmed, Ashwini Maratha, Aisha Qasim Butt, Enda Shevlin and Sinead M. Miggin This information is current as of September 27, 2021. J Immunol 2013; 190:2217-2228; Prepublished online 30 January 2013; doi: 10.4049/jimmunol.1201630 http://www.jimmunol.org/content/190/5/2217 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2013/01/30/jimmunol.120163 Material 0.DC1 References This article cites 57 articles, 20 of which you can access for free at: http://www.jimmunol.org/ http://www.jimmunol.org/content/190/5/2217.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 27, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology TRIF-Mediated TLR3 and TLR4 Signaling Is Negatively Regulated by ADAM15 Suaad Ahmed, Ashwini Maratha, Aisha Qasim Butt, Enda Shevlin, and Sinead M. -

ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease
International Journal of Molecular Sciences Review ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease Francesca Tosetti 1,* , Massimo Alessio 2, Alessandro Poggi 1,† and Maria Raffaella Zocchi 3,† 1 Molecular Oncology and Angiogenesis Unit, IRCCS Ospedale Policlinico S. Martino Largo R. Benzi 10, 16132 Genoa, Italy; [email protected] 2 Proteome Biochemistry, IRCCS San Raffaele Scientific Institute, 20132 Milan, Italy; [email protected] 3 Division of Immunology, Transplants and Infectious Diseases, IRCCS San Raffaele Scientific Institute, 20132 Milan, Italy; [email protected] * Correspondence: [email protected] † These authors contributed equally to this work as last author. Abstract: Enzymes, once considered static molecular machines acting in defined spatial patterns and sites of action, move to different intra- and extracellular locations, changing their function. This topological regulation revealed a close cross-talk between proteases and signaling events involving post-translational modifications, membrane tyrosine kinase receptors and G-protein coupled recep- tors, motor proteins shuttling cargos in intracellular vesicles, and small-molecule messengers. Here, we highlight recent advances in our knowledge of regulation and function of A Disintegrin And Metalloproteinase (ADAM) endopeptidases at specific subcellular sites, or in multimolecular com- plexes, with a special focus on ADAM10, and tumor necrosis factor-α convertase (TACE/ADAM17), since these two enzymes belong to the same family, share selected substrates and bioactivity. We will discuss some examples of ADAM10 activity modulated by changing partners and subcellular compartmentalization, with the underlying hypothesis that restraining protease activity by spatial Citation: Tosetti, F.; Alessio, M.; segregation is a complex and powerful regulatory tool. -

Cell-Autonomous FLT3L Shedding Via ADAM10 Mediates Conventional Dendritic Cell Development in Mouse Spleen
Cell-autonomous FLT3L shedding via ADAM10 mediates conventional dendritic cell development in mouse spleen Kohei Fujitaa,b,1, Svetoslav Chakarovc,1, Tetsuro Kobayashid, Keiko Sakamotod, Benjamin Voisind, Kaibo Duanc, Taneaki Nakagawaa, Keisuke Horiuchie, Masayuki Amagaib, Florent Ginhouxc, and Keisuke Nagaod,2 aDepartment of Dentistry and Oral Surgery, Keio University School of Medicine, Tokyo 160-8582, Japan; bDepartment of Dermatology, Keio University School of Medicine, Tokyo 160-8582, Japan; cSingapore Immunology Network, Agency for Science, Technology and Research, Biopolis, 138648 Singapore; dDermatology Branch, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, MD 20892; and eDepartment of Orthopedic Surgery, National Defense Medical College, Tokorozawa 359-8513, Japan Edited by Kenneth M. Murphy, Washington University School of Medicine, St. Louis, MO, and approved June 10, 2019 (received for review November 4, 2018) Conventional dendritic cells (cDCs) derive from bone marrow (BM) intocDC1sorcDC2stakesplaceintheBM(3),andthese precursors that undergo cascades of developmental programs to pre-cDC1s and pre-cDC2s ultimately differentiate into cDC1s terminally differentiate in peripheral tissues. Pre-cDC1s and pre- and cDC2s after migrating to nonlymphoid and lymphoid tissues. + + cDC2s commit in the BM to each differentiate into CD8α /CD103 cDCs are short-lived, and their homeostatic maintenance relies + cDC1s and CD11b cDC2s, respectively. Although both cDCs rely on on constant replenishment from the BM precursors (5). The cy- the cytokine FLT3L during development, mechanisms that ensure tokine Fms-related tyrosine kinase 3 ligand (FLT3L) (12), by cDC accessibility to FLT3L have yet to be elucidated. Here, we gen- signaling through its receptor FLT3 expressed on DC precursors, erated mice that lacked a disintegrin and metalloproteinase (ADAM) is essential during the development of DCs (7, 13). -

Expression of the Disintegrin Metalloprotease, ADAM-10, In
314 Vol. 10, 314–323, January 1, 2004 Clinical Cancer Research Expression of the Disintegrin Metalloprotease, ADAM-10, in Prostate Cancer and Its Regulation by Dihydrotestosterone, Insulin-Like Growth Factor I, and Epidermal Growth Factor in the Prostate Cancer Cell Model LNCaP Daniel R. McCulloch,1 Pascal Akl,1 Conclusions: This study describes for the first time Hemamali Samaratunga,2 Adrian C. Herington,1 the expression, regulation, and cellular localization of and Dimitri M. Odorico1 ADAM-10 protein in PCa. The regulation and membrane 1 localization of ADAM-10 support our hypothesis that Hormone-Dependent Cancer Program, School of Life Sciences, ADAM-10 has a role in extracellular matrix maintenance Queensland University of Technology, Brisbane, Queensland, Australia, and 2Sullivan Nicolaides Pathology, Brisbane, Queensland, and cell invasion, although the potential role of nuclear Australia ADAM-10 is not yet known. INTRODUCTION ABSTRACT The disintegrin metalloproteases ADAMs, like the matrix Purpose: The disintegrin metalloprotease ADAM-10 is metalloproteinases (MMPs), are members of the metzincin a multidomain metalloprotease that is potentially significant (zinc-dependent metalloprotease) superfamily. To date, more in tumor progression due to its extracellular matrix-degrad- than 30 ADAMs have been characterized (1), some of which are ing properties. Previously, ADAM-10 mRNA was detected involved in diverse biological functions such as fertilization, in prostate cancer (PCa) cell lines; however, the presence of neurogenesis (2, 3), and the ectodomain shedding of growth ADAM-10 protein and its cellular localization, regulation, factors such as amyloid precursor protein and tumor necrosis and role have yet to be described. We hypothesized that factor ␣ (4, 5). -
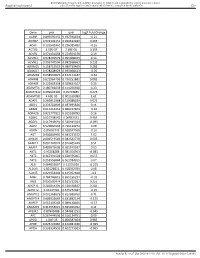
Gene Pval Qval Log2 Fold Change AAMP 0.895690332 0.952598834
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut Gene pval qval Log2 Fold Change AAMP 0.895690332 0.952598834 -0.21 ABI3BP 0.002302151 0.020612283 0.465 ACHE 0.103542461 0.296385483 -0.16 ACTG2 2.99E-07 7.68E-05 3.195 ACVR1 0.071431098 0.224504378 0.19 ACVR1C 0.978209579 0.995008423 0.14 ACVRL1 0.006747504 0.042938663 0.235 ADAM15 0.158715519 0.380719469 0.285 ADAM17 0.978208929 0.995008423 -0.05 ADAM28 0.038932876 0.152174187 -0.62 ADAM8 0.622964796 0.790251882 0.085 ADAM9 0.122003358 0.329623107 0.25 ADAMTS1 0.180766659 0.414256926 0.23 ADAMTS12 0.009902195 0.05703885 0.425 ADAMTS8 4.60E-05 0.001169089 1.61 ADAP1 0.269811968 0.519388039 0.075 ADD1 0.233702809 0.487695826 0.11 ADM2 0.012213453 0.066227879 -0.36 ADRA2B 0.822777921 0.915518785 0.16 AEBP1 0.010738542 0.06035531 0.465 AGGF1 0.117946691 0.320915024 -0.095 AGR2 0.529860903 0.736120272 0.08 AGRN 0.85693743 0.928047568 -0.16 AGT 0.006849995 0.043233572 1.02 AHNAK 0.006519543 0.042542779 0.605 AKAP12 0.001747074 0.016405449 0.51 AKAP2 0.409929603 0.665919397 0.05 AKT1 0.95208288 0.985354963 -0.085 AKT2 0.367391504 0.620376005 0.055 AKT3 0.253556844 0.501934205 0.07 ALB 0.064833867 0.21195036 -0.315 ALDOA 0.83128831 0.918352939 0.08 ALOX5 0.029954404 0.125352668 -0.3 AMH 0.784746815 0.895196237 -0.03 ANG 0.050500474 0.181732067 0.255 ANGPT1 0.281853305 0.538528647 0.285 ANGPT2 0.43147281 0.675272487 -0.15 ANGPTL2 0.001368876 0.013688762 0.71 ANGPTL4 0.686032669 0.831882134 -0.175 ANPEP 0.019103243 0.089148466 -0.57 ANXA2P2 0.412553021 0.665966092 0.11 AP1M2 0.87843088 0.944681253 -0.045 APC 0.267444505 0.516134751 0.09 APOD 1.04E-05 0.000587404 0.985 APOE 0.023722987 0.104981036 -0.395 APOH 0.336334555 0.602273505 -0.065 Sundar R, et al. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like,