Activation of the Murine EP3 Receptor for PGE2 Inhibits Camp Production and Promotes Platelet Aggregation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cox Inhibitors and Thromboregulation
CLINICAL IMPLICATIONS OF BASIC RESEARCH Clinical Implications shown the importance of eicosanoids in preserving of Basic Research the dynamic balance among thrombosis, hemostasis, and the fluidity of blood. Recently, Cheng et al.1 presented compelling evi- dence that cell–cell interactions, principally between platelets and endothelial cells, that are mediated by ei- COX INHIBITORS cosanoids have a role in thrombosis. Using genetical- AND THROMBOREGULATION ly engineered mice that either overexpressed or lacked essential components of the eicosanoid pathway — ROM a historical perspective, there is perhaps no namely, receptors for prostacyclin (a platelet inhibitor Fmore interesting therapeutic saga than that of as- and vasodilator) or thromboxane A2 (a platelet agonist pirin, which began as a folk remedy, distilled from wil- and vasoconstrictor) — they found that the response low bark, and became a lifesaving preventive treatment of the intima of carotid vessels to mechanical injury is for ischemic cardiovascular disease. Aspirin primarily exuberant and leads to obstruction in mice lacking the inhibits the cyclooxygenase (COX)-dependent synthe- prostacyclin receptor. However, this response is muted sis of eicosanoids, which are the end products of me- in mice lacking the thromboxane A2 receptor or both tabolism of essential fatty acids and include prosta- receptors. In mice lacking the prostacyclin receptor cyclin and thromboxane A2. Numerous studies have (a defect that mimics the effects of COX-2–selective Endothelial cell Resting platelet Thromboxane A2 Soluble Stimulation CD39 Arachidonic acid Stimulated platelet COX inhibitor ATP and ADP Prostacyclin Nitric oxide AMP Carbon monoxide CD39 Resting platelet Figure 1. Effect on Platelet Reactivity of the Eicosanoids Thromboxane A2 and Prostacyclin, the Biologic Gases Nitric Oxide and Carbon Monoxide, and the Ectonucleotidase CD39. -

Download Product Insert (PDF)
Product Information U-46619 Item No. 16450 CAS Registry No.: 56985-40-1 Formal Name: 9,11-dideoxy-9α,11α-methanoepoxy-prosta- 5Z,13E-dien-1-oic acid Synonym: 9,11-dideoxy-9α,11α-methanoepoxy COOH Prostaglandin F2α MF: C21H34O4 O FW: 350.5 Purity: ≥98% OH Stability: ≥2 years at -20°C Supplied as: A solution in methyl acetate Laboratory Procedures For long term storage, we suggest that U-46619 be stored as supplied at -20°C. It will be stable for at least two years. U-46619 is supplied as a solution in methyl acetate. To change the solvent, simply evaporate the methyl acetate under a gentle stream of nitrogen and immediately add the solvent of choice. Solvents such as DMSO, ethanol, and dimethyl formamide purged with an inert gas can be used. The solubility of U-46619 in these solvents is aproximately 100 mg/ml. Further dilutions of the stock solution into aqueous buffers or isotonic saline should be made prior to performing biological experiments. Ensure that the residual amount of organic solvent is insignificant since organic solvents may have physiological effects at low concentrations. If an organic solvent-free solution of U-46619 is needed, it can be prepared by evaporating the methyl acetate and directly dissolving the neat oil in aqueous buffers. The solubility of U-46619 in PBS (pH 7.2) is approximately 2 mg/ml. We do not recommend storing the aqueous solution for more than one day. 1 U-46619 is a stable analog of the endoperoxide prostaglandin H2, and a TP receptor agonist. -

Endogenous Biosynthesis of Prostacyclin and Thromboxane and Platelet Function During Chronic Administration of Aspirin in Man
Endogenous biosynthesis of prostacyclin and thromboxane and platelet function during chronic administration of aspirin in man. G A FitzGerald, … , J A Lawson, A R Brash J Clin Invest. 1983;71(3):676-688. https://doi.org/10.1172/JCI110814. Research Article To assess the pharmacologic effects of aspirin on endogenous prostacyclin and thromboxane biosynthesis, 2,3-dinor-6- keto PGF1 alpha (PGI-M) and 2,3-dinor-thromboxane B2 (Tx-M) were measured in urine by mass spectrometry during continuing administration of aspirin. To define the relationship of aspirin intake to endogenous prostacyclin biosynthesis, sequential urines were initially collected in individuals prior to, during, and subsequent to administration of aspirin. Despite inter- and intra-individual variations, PGI-M excretion was significantly reduced by aspirin. However, full mass spectral identification confirmed continuing prostacyclin biosynthesis during aspirin therapy. Recovery of prostacyclin biosynthesis was incomplete 5 d after drug administration was discontinued. To relate aspirin intake to indices of thromboxane biosynthesis and platelet function, volunteers received 20 mg aspirin daily followed by 2,600 mg aspirin daily, each dose for 7 d in sequential weeks. Increasing aspirin dosage inhibited Tx-M excretion from 70 to 98% of pretreatment control values; platelet TxB2 formation from 4.9 to 0.5% and further inhibited platelet function. An extended study was performed to relate aspirin intake to both thromboxane and prostacyclin generation over a wide range of doses. Aspirin, in the range of 20 to 325 mg/d, resulted in a dose-dependent decline in both Tx-M and PGI-M excretion. At doses of 325-2,600 mg/d Tx-M excretion ranged from 5 to 3% of control values while PGI-M remained at 37-23% of control. -

Challenging the FDA Black Box Warning for High Aspirin Dose with Ticagrelor in Patients with Diabetes James J
PERSPECTIVES IN DIABETES Challenging the FDA Black Box Warning for High Aspirin Dose With Ticagrelor in Patients With Diabetes James J. DiNicolantonio and Victor L. Serebruany – Ticagrelor, a novel reversible antiplatelet agent, has a Food and ASA 300 325 mg (53.6%) in the U.S. compared with the Drug Administration (FDA) black box warning to avoid mainte- rest of the world (1.7%) was the only factor to explain nance doses of aspirin (ASA) .100 mg/daily. This restriction is (out of 37 variables explored) the regional interaction based on the hypothesis that ASA doses .100 mg somehow de- that ticagrelor was less effective and potentially more creased ticagrelor’s benefit in the Platelet Inhibition and Patient harmful than clopidogrel (2). However, this interaction Outcomes (PLATO) U.S. cohort. However, these data are highly was not significant, is highly postrandomized, comes postrandomized, come from a very small subgroup in PLATO from a very small subgroup of PLATO, and makes no (57% of patients in the U.S. site), and make no biological sense. biological sense (3). Moreover, the ticagrelor-ASA interaction was not significant by any multivariate Cox regression analyses. The Complete Re- sponse Review for ticagrelor indicates that for U.S. PLATO patients, an ASA dose .300 mg was not a significant interaction TICAGRELOR-ASA HYPOTHESIS for vascular outcomes. In the ticagrelor-ASA .300 mg cohort, all- The claimed ticagrelor-ASA interaction states that ticagrelor cause and vascular mortality were not significantly increased fi – P plus low-dose ASA is bene cial, whereas increasing doses (hazard ratio [HR] 1.27 [95% CI 0.84 1.93], = 0.262 and 1.39 of ASA in combination with ticagrelor produces adverse [0.87–2.2], P = 0.170), respectively. -

GTH 2021 State of the Art—Cardiac Surgery: the Perioperative Management of Heparin-Induced Thrombocytopenia in Cardiac Surgery
Review Article 59 GTH 2021 State of the Art—Cardiac Surgery: The Perioperative Management of Heparin-Induced Thrombocytopenia in Cardiac Surgery Laura Ranta1 Emmanuelle Scala1 1 Department of Anesthesiology, Cardiothoracic and Vascular Address for correspondence Emmanuelle Scala, MD, Centre Anesthesia, Lausanne University Hospital (CHUV), Lausanne, Hospitalier Universitaire Vaudois, Rue du Bugnon 46, BH 05/300, 1011 Switzerland Lausanne, Suisse, Switzerland (e-mail: [email protected]). Hämostaseologie 2021;41:59–62. Abstract Heparin-induced thrombocytopenia (HIT) is a severe, immune-mediated, adverse drug Keywords reaction that paradoxically induces a prothrombotic state. Particularly in the setting of ► Heparin-induced cardiac surgery, where full anticoagulation is required during cardiopulmonary bypass, thrombocytopenia the management of HIT can be highly challenging, and requires a multidisciplinary ► cardiac surgery approach. In this short review, the different perioperative strategies to run cardiopul- ► state of the art monary bypass will be summarized. Introduction genicity of the antibodies and is diagnostic for HIT. The administration of heparin to a patient with circulating Heparin-induced thrombocytopenia (HIT) is a severe, im- pathogenic HITabs puts the patient at immediate risk of mune-mediated, adverse drug reaction that paradoxically severe thrombotic complications. induces a prothrombotic state.1,2 Particularly in the setting The time course of HIT can be divided into four distinct of cardiac surgery, where full anticoagulation is required phases.6 Acute HIT is characterized by thrombocytopenia during cardiopulmonary bypass (CPB), the management of and/or thrombosis, the presence of HITabs, and confirma- HIT can be highly challenging, and requires a multidisciplin- tion of their platelet activating capacity by a functional ary approach. -

Inverse Agonism of SQ 29,548 and Ramatroban on Thromboxane A2 Receptor
Inverse Agonism of SQ 29,548 and Ramatroban on Thromboxane A2 Receptor Raja Chakraborty1,3, Rajinder P. Bhullar1, Shyamala Dakshinamurti2,3, John Hwa4, Prashen Chelikani1,2,3* 1 Department of Oral Biology, University of Manitoba, Winnipeg, Manitoba, Canada, 2 Departments of Pediatrics, Physiology, University of Manitoba, Winnipeg, Manitoba, Canada, 3 Biology of Breathing Group- Manitoba Institute of Child Health, Winnipeg, Manitoba, Canada, 4 Department of Internal Medicine (Cardiology), Cardiovascular Research Center, Yale University School of Medicine, New Haven, Connecticut, United States of America Abstract G protein-coupled receptors (GPCRs) show some level of basal activity even in the absence of an agonist, a phenomenon referred to as constitutive activity. Such constitutive activity in GPCRs is known to have important pathophysiological roles in human disease. The thromboxane A2 receptor (TP) is a GPCR that promotes thrombosis in response to binding of the prostanoid, thromboxane A2. TP dysfunction is widely implicated in pathophysiological conditions such as bleeding disorders, hypertension and cardiovascular disease. Recently, we reported the characterization of a few constitutively active mutants (CAMs) in TP, including a genetic variant A160T. Using these CAMs as reporters, we now test the inverse agonist properties of known antagonists of TP, SQ 29,548, Ramatroban, L-670596 and Diclofenac, in HEK293T cells. Interestingly, SQ 29,548 reduced the basal activity of both, WT-TP and the CAMs while Ramatroban was able to reduce the basal activity of only the CAMs. Diclofenac and L-670596 showed no statistically significant reduction in basal activity of WT-TP or CAMs. To investigate the role of these compounds on human platelet function, we tested their effects on human megakaryocyte based system for platelet activation. -

Role of Proaggregatory and Antiaggregatory Prostaglandins in Hemostasis
Role of proaggregatory and antiaggregatory prostaglandins in hemostasis. Studies with combined thromboxane synthase inhibition and thromboxane receptor antagonism. P Gresele, … , G Pieters, J Vermylen J Clin Invest. 1987;80(5):1435-1445. https://doi.org/10.1172/JCI113223. Research Article Thromboxane synthase inhibition can lead to two opposing effects: accumulation of proaggregatory cyclic endoperoxides and increased formation of antiaggregatory PGI2 and PGD2. The elimination of the effects of the cyclic endoperoxides by an endoperoxide-thromboxane A2 receptor antagonist should enhance the inhibition of hemostasis by thromboxane synthase blockers. We have carried out a series of double-blind, placebo-controlled, crossover studies in healthy volunteers to check if this hypothesis may be operative in vivo in man. In a first study, in 10 healthy male volunteers, the combined administration of the thromboxane receptor antagonist BM 13.177 and the thromboxane synthase inhibitor dazoxiben gave stronger inhibition of platelet aggregation and prolonged the bleeding time more than either drug alone. In a second study, in 10 different healthy male volunteers, complete inhibition of cyclooxygenase with indomethacin reduced the prolongation of the bleeding time by the combination BM 13.177 plus dazoxiben. In a third study, in five volunteers, selective cumulative inhibition of platelet TXA2 synthesis by low-dose aspirin inhibited platelet aggregation and prolonged the bleeding time less than the combination BM 13.177 plus dazoxiben. In vitro, in -
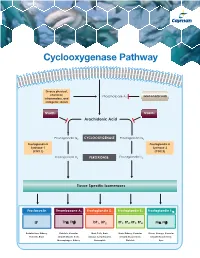
Cyclooxygenase Pathway
Cyclooxygenase Pathway Diverse physical, chemical, Phospholipase A Glucocorticoids inflammatory, and 2 mitogenic stimuli NSAIDs NSAIDs Arachidonic Acid Prostaglandin G2 CYCLOOXYGENASE Prostaglandin G2 Prostaglandin H Prostaglandin H Synthase-1 Synthase-2 (COX 1) (COX 2) Prostaglandin H2 PEROXIDASE Prostaglandin H2 Tissue Specific Isomerases Prostacyclin Thromboxane A2 Prostaglandin D2 Prostaglandin E2 Prostaglandin F2α IP TPα, TPβ DP1, DP2 EP1, EP2, EP3, EP4 FPα, FPβ Endothelium, Kidney, Platelets, Vascular Mast Cells, Brain, Brain, Kidney, Vascular Uterus, Airways, Vascular Platelets, Brain Smooth Muscle Cells, Airways, Lymphocytes, Smooth Muscle Cells, Smooth Muscle Cells, Macrophages, Kidney Eosinophils Platelets Eyes Prostacyclin Item No. Product Features Prostacyclin (Prostaglandin I2; PGI2) is formed from arachidonic acid primarily in the vascular endothelium and renal cortex by sequential 515211 6-keto • Sample Types: Culture Medium | Plasma Prostaglandin • Measure 6-keto PGF levels down to 6 pg/ml activities of COX and prostacyclin synthase. PGI2 is non-enzymatically 1α F ELISA Kit • Incubation : 18 hours | Development: 90-120 minutes | hydrated to 6-keto PGF1α (t½ = 2-3 minutes), and then quickly converted 1α Read: Colorimetric at 405-420 nm to the major metabolite, 2,3-dinor-6-keto PGF1α (t½= 30 minutes). Prostacyclin was once thought to be a circulating hormone that regulated • Assay 24 samples in triplicate or 36 samples in duplicate platelet-vasculature interactions, but the rate of secretion into circulation • NOTE: A portion of urinary 6-keto PGF1α is of renal origin coupled with the short half-life indicate that prostacyclin functions • NOTE : It has been found that normal plasma levels of 6-keto PGF may be low locally. -

Vasoactive Responses of U46619, PGF2 , Latanoprost, and Travoprost
Vasoactive Responses of U46619, PGF2␣, Latanoprost, and Travoprost in Isolated Porcine Ciliary Arteries Ineta Vysniauskiene, Reto Allemann, Josef Flammer, and Ivan O. Haefliger PURPOSE. To compare the vasoactive properties of the prosta- these responses can be modulated by SQ 29548 (TP-receptor noids U46619 (thromboxane A2 analogue), prostaglandin F2␣ antagonist) or AL-8810 (FP-receptor antagonist). (PGF2␣), latanoprost free acid, and travoprost free acid in isolated porcine ciliary arteries. METHODS. In a myograph system (isometric force measure- MATERIAL AND METHODS ment), quiescent vessels were exposed (cumulatively) to U46619, PGF , latanoprost, or travoprost (0.1 nM–0.1 mM). 2␣ Vessels Preparation Experiments were also conducted in the presence of SQ 29548 (TP-receptor antagonist; 3–10 M) or AL-8810 (FP-receptor Porcine eyes were obtained from an abattoir immediately after death. antagonist; 3–30 M). Contractions were expressed as the In cold modified Krebs-Ringer solution (NaCl 118 mM, KCl 4.7 mM, percentage of 100 mM potassium chloride-induced contrac- CaCl2 2.5 mM, K2PO4 1.2 mM, MgSO4 1.2 mM, NaHCO3 25 mM, tions. glucose 11.1 mM, and EDTA 0.026 mM), ciliary arteries were dissected 5 RESULTS. In quiescent vessels, contractions (concentration–re- and cut into 2-mm segments. In an organ chamber, two 45- m Ϯ tungsten wires were passed through the vessel’s lumen and attached to sponse curves) induced by (0.1 mM) PGF2␣ (87.9% 3.5%), U46619 (66.7% Ϯ 4.1%), and latanoprost (62.9% Ϯ 3.6%) were a force transducer for isometric force measurements (Myo-Interface; JP more pronounced (P Յ 0.001) than those induced by tra- Trading, Aarhus, Denmark). -

Effect of Prostanoids on Human Platelet Function: an Overview
International Journal of Molecular Sciences Review Effect of Prostanoids on Human Platelet Function: An Overview Steffen Braune, Jan-Heiner Küpper and Friedrich Jung * Institute of Biotechnology, Molecular Cell Biology, Brandenburg University of Technology, 01968 Senftenberg, Germany; steff[email protected] (S.B.); [email protected] (J.-H.K.) * Correspondence: [email protected] Received: 23 October 2020; Accepted: 23 November 2020; Published: 27 November 2020 Abstract: Prostanoids are bioactive lipid mediators and take part in many physiological and pathophysiological processes in practically every organ, tissue and cell, including the vascular, renal, gastrointestinal and reproductive systems. In this review, we focus on their influence on platelets, which are key elements in thrombosis and hemostasis. The function of platelets is influenced by mediators in the blood and the vascular wall. Activated platelets aggregate and release bioactive substances, thereby activating further neighbored platelets, which finally can lead to the formation of thrombi. Prostanoids regulate the function of blood platelets by both activating or inhibiting and so are involved in hemostasis. Each prostanoid has a unique activity profile and, thus, a specific profile of action. This article reviews the effects of the following prostanoids: prostaglandin-D2 (PGD2), prostaglandin-E1, -E2 and E3 (PGE1, PGE2, PGE3), prostaglandin F2α (PGF2α), prostacyclin (PGI2) and thromboxane-A2 (TXA2) on platelet activation and aggregation via their respective receptors. Keywords: prostacyclin; thromboxane; prostaglandin; platelets 1. Introduction Hemostasis is a complex process that requires the interplay of multiple physiological pathways. Cellular and molecular mechanisms interact to stop bleedings of injured blood vessels or to seal denuded sub-endothelium with localized clot formation (Figure1). -

Antiplatelet Effects of Prostacyclin Analogues: Which One to Choose in Case of Thrombosis Or Bleeding? Sylwester P
INTERVENTIONAL CARDIOLOGY Cardiology Journal 20XX, Vol. XX, No. X, XXX–XXX DOI: 10.5603/CJ.a2020.0164 Copyright © 20XX Via Medica REVIEW ARTICLE ISSN 1897–5593 eISSN 1898–018X Antiplatelet effects of prostacyclin analogues: Which one to choose in case of thrombosis or bleeding? Sylwester P. Rogula1*, Hubert M. Mutwil1*, Aleksandra Gąsecka1, Marcin Kurzyna2, Krzysztof J. Filipiak1 11st Chair and Department of Cardiology, Medical University of Warsaw, Poland 2Department of Pulmonary Circulation, Thromboembolic Diseases and Cardiology, Center of Postgraduate Education Medical, European Health Center Otwock, Poland Abstract Prostacyclin and analogues are successfully used in the treatment of pulmonary arterial hypertension (PAH) due to their vasodilatory effect on pulmonary arteries. Besides vasodilatory effect, prostacyclin analogues inhibit platelets, but their antiplatelet effect is not thoroughly established. The antiplatelet effect of prostacyclin analogues may be beneficial in case of increased risk of thromboembolic events, or undesirable in case of increased risk of bleeding. Since prostacyclin and analogues differ regarding their potency and form of administration, they might also inhibit platelets to a different extent. This review summarizes the recent evidence on the antiplatelet effects of prostacyclin and analogue in the treatment of PAH, this is important to consider when choosing the optimal treatment regimen in tailoring to an individual patients’ needs. (Cardiol J 20XX; XX, X: xx–xx) Key words: prostacyclin analogues, pulmonary arterial hypertension, platelets, antiplatelet effect, thrombosis, bleeding Introduction tiproliferative effects [4]. The main indication for PGI2 and analogues is advanced pulmonary arterial Since 1935 when prostaglandin was isolated hypertension (PAH) and peripheral vascular disor- for the first time [1], many scientists have focused ders [5]. -

Multi-Functionality of Proteins Involved in GPCR and G Protein Signaling: Making Sense of Structure–Function Continuum with In
Cellular and Molecular Life Sciences (2019) 76:4461–4492 https://doi.org/10.1007/s00018-019-03276-1 Cellular andMolecular Life Sciences REVIEW Multi‑functionality of proteins involved in GPCR and G protein signaling: making sense of structure–function continuum with intrinsic disorder‑based proteoforms Alexander V. Fonin1 · April L. Darling2 · Irina M. Kuznetsova1 · Konstantin K. Turoverov1,3 · Vladimir N. Uversky2,4 Received: 5 August 2019 / Revised: 5 August 2019 / Accepted: 12 August 2019 / Published online: 19 August 2019 © Springer Nature Switzerland AG 2019 Abstract GPCR–G protein signaling system recognizes a multitude of extracellular ligands and triggers a variety of intracellular signal- ing cascades in response. In humans, this system includes more than 800 various GPCRs and a large set of heterotrimeric G proteins. Complexity of this system goes far beyond a multitude of pair-wise ligand–GPCR and GPCR–G protein interactions. In fact, one GPCR can recognize more than one extracellular signal and interact with more than one G protein. Furthermore, one ligand can activate more than one GPCR, and multiple GPCRs can couple to the same G protein. This defnes an intricate multifunctionality of this important signaling system. Here, we show that the multifunctionality of GPCR–G protein system represents an illustrative example of the protein structure–function continuum, where structures of the involved proteins represent a complex mosaic of diferently folded regions (foldons, non-foldons, unfoldons, semi-foldons, and inducible foldons). The functionality of resulting highly dynamic conformational ensembles is fne-tuned by various post-translational modifcations and alternative splicing, and such ensembles can undergo dramatic changes at interaction with their specifc partners.