Experimental and Theoretical Comparison of Actinide and Lanthanide Bonding in M[N(EPR2)2]3 Complexes (M ) U, Pu, La, Ce; E ) S, Se, Te; R ) Ph, Ipr, H)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Additional Teacher Background Chapter 4 Lesson 3, P. 295
Additional Teacher Background Chapter 4 Lesson 3, p. 295 What determines the shape of the standard periodic table? One common question about the periodic table is why it has its distinctive shape. There are actu- ally many different ways to represent the periodic table including circular, spiral, and 3-D. But in most cases, it is shown as a basically horizontal chart with the elements making up a certain num- ber of rows and columns. In this view, the table is not a symmetrical rectangular chart but seems to have steps or pieces missing. The key to understanding the shape of the periodic table is to recognize that the characteristics of the atoms themselves and their relationships to one another determine the shape and patterns of the table. ©2011 American Chemical Society Middle School Chemistry Unit 303 A helpful starting point for explaining the shape of the periodic table is to look closely at the structure of the atoms themselves. You can see some important characteristics of atoms by look- ing at the chart of energy level diagrams. Remember that an energy level is a region around an atom’s nucleus that can hold a certain number of electrons. The chart shows the number of energy levels for each element as concentric shaded rings. It also shows the number of protons (atomic number) for each element under the element’s name. The electrons, which equal the number of protons, are shown as dots within the energy levels. The relationship between atomic number, energy levels, and the way electrons fill these levels determines the shape of the standard periodic table. -

Actinide Ground-State Properties-Theoretical Predictions
Actinide Ground-State Properties Theoretical predictions John M. Wills and Olle Eriksson electron-electron correlations—the electronic energy of the ground state of or nearly fifty years, the actinides interactions among the 5f electrons and solids, molecules, and atoms as a func- defied the efforts of solid-state between them and other electrons—are tional of electron density. The DFT Ftheorists to understand their expected to affect the bonding. prescription has had such a profound properties. These metals are among Low-symmetry crystal structures, impact on basic research in both the most complex of the long-lived relativistic effects, and electron- chemistry and solid-state physics that elements, and in the solid state, they electron correlations are very difficult Walter Kohn, its main inventor, was display some of the most unusual to treat in traditional electronic- one of the recipients of the 1998 behaviors of any series in the periodic structure calculations of metals and, Nobel Prize in Chemistry. table. Very low melting temperatures, until the last decade, were outside the In general, it is not possible to apply large anisotropic thermal-expansion realm of computational ability. And DFT without some approximation. coefficients, very low symmetry crystal yet, it is essential to treat these effects But many man-years of intense research structures, many solid-to-solid phase properly in order to understand the have yielded reliable approximate transitions—the list is daunting. Where physics of the actinides. Electron- expressions for the total energy in does one begin to put together an electron correlations are important in which all terms, except for a single- understanding of these elements? determining the degree to which 5f particle kinetic-energy term, can be In the last 10 years, together with electrons are localized at lattice sites. -
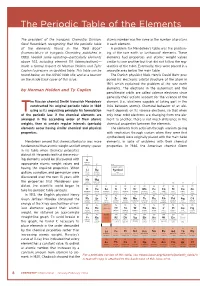
The Periodic Table of the Elements
The Periodic Table of the Elements The president of the Inorganic Chemistry Division, atomic number was the same as the number of protons Gerd Rosenblatt, recognizing that the periodic table in each element. of the elements found in the “Red Book” A problem for Mendeleev’s table was the position- (Nomenclature of Inorganic Chemistry, published in ing of the rare earth or lanthanoid* elements. These 1985) needed some updating—particularly elements elements had properties and atomic weight values above 103, including element 110 (darmstadtium)— similar to one another but that did not follow the reg- made a formal request to Norman Holden and Tyler ularities of the table. Eventually, they were placed in a Coplen to prepare an updated table. This table can be separate area below the main table. found below, on the IUPAC Web site, and as a tear-off The Danish physicist Niels Henrik David Bohr pro- on the inside back cover of this issue. posed his electronic orbital structure of the atom in 1921, which explained the problem of the rare earth by Norman Holden and Ty Coplen elements. The electrons in the outermost and the penultimate orbits are called valence electrons since generally their actions account for the valence of the he Russian chemist Dmitri Ivanovich Mendeleev element (i.e., electrons capable of taking part in the constructed his original periodic table in 1869 links between atoms). Chemical behavior of an ele- Tusing as its organizing principle his formulation ment depends on its valence electrons, so that when of the periodic law: if the chemical elements are only inner orbit electrons are changing from one ele- arranged in the ascending order of their atomic ment to another, there is not much difference in the weights, then at certain regular intervals (periods) chemical properties between the elements. -

ELECTRONEGATIVITY D Qkq F=
ELECTRONEGATIVITY The electronegativity of an atom is the attracting power that the nucleus has for it’s own outer electrons and those of it’s neighbours i.e. how badly it wants electrons. An atom’s electronegativity is determined by Coulomb’s Law, which states, “ the size of the force is proportional to the size of the charges and inversely proportional to the square of the distance between them”. In symbols it is represented as: kq q F = 1 2 d 2 where: F = force (N) k = constant (dependent on the medium through which the force is acting) e.g. air q1 = charge on an electron (C) q2 = core charge (C) = no. of protons an outer electron sees = no. of protons – no. of inner shell electrons = main Group Number d = distance of electron from the nucleus (m) Examples of how to calculate the core charge: Sodium – Atomic number 11 Electron configuration 2.8.1 Core charge = 11 (no. of p+s) – 10 (no. of inner e-s) +1 (Group i) Chlorine – Atomic number 17 Electron configuration 2.8.7 Core charge = 17 (no. of p+s) – 10 (no. of inner e-s) +7 (Group vii) J:\Sciclunm\Resources\Year 11 Chemistry\Semester1\Notes\Atoms & The Periodic Table\Electronegativity.doc Neon holds onto its own electrons with a core charge of +8, but it can’t hold any more electrons in that shell. If it was to form bonds, the electron must go into the next shell where the core charge is zero. Group viii elements do not form any compounds under normal conditions and are therefore given no electronegativity values. -

Is Selenium a Metal, Non-Metal Or Metalloid?
Is Selenium a metal, non-metal or metalloid? Abstract Is selenium(Se) a metal, non-metal, or a metalloid? There are various public opinions circulating around it. Since a long time from now, there are a lot of voices discussing this. Even until now, there is still no consensus about it. So, in this project, we are trying to find out whether selenium is a non-metal, metal or metalloids base on its physical and chemical properties which could be studied in the secondary school combining with the other information from the internet. Principles and hypothesis Studied from the secondary school chemistry, the general properties of metals include being good conductors of heat and electricity, having high melting and boiling points. Non-metals generally have a lower melting point and boiling point than metals and they being poor conductors of heat and electricity, etc. And the physical properties of metalloids are having extremely high melting/ boiling point, and having fair electrical conductivity. On the other hand, the oxides of metal are generally basic whereas the oxide of non-metals and metalloids are generally acidic. We will define selenium’s chemical category based on the above properties. Besides, we would like to introduce the concept of displacement reaction in studying the chemical properties of the chalcogens(e.g. sulphur(S) and selenium(Se)), especially selenium such rarely mentioned element. We can assume selenium is a metal if selenium could displace a metal oxide or a metal could displace selenium (IV) oxide. If selenium is a non-metal, its oxide could be displaced by sulphur which is supposed to be more reactive than selenium in the chalcogen group. -

Periodic Table of the Elements Notes
Periodic Table of the Elements Notes Arrangement of the known elements based on atomic number and chemical and physical properties. Divided into three basic categories: Metals (left side of the table) Nonmetals (right side of the table) Metalloids (touching the zig zag line) Basic Organization by: Atomic structure Atomic number Chemical and Physical Properties Uses of the Periodic Table Useful in predicting: chemical behavior of the elements trends properties of the elements Atomic Structure Review: Atoms are made of protons, electrons, and neutrons. Elements are atoms of only one type. Elements are identified by the atomic number (# of protons in nucleus). Energy Levels Review: Electrons are arranged in a region around the nucleus called an electron cloud. Energy levels are located within the cloud. At least 1 energy level and as many as 7 energy levels exist in atoms Energy Levels & Valence Electrons Energy levels hold a specific amount of electrons: 1st level = up to 2 2nd level = up to 8 3rd level = up to 8 (first 18 elements only) The electrons in the outermost level are called valence electrons. Determine reactivity - how elements will react with others to form compounds Outermost level does not usually fill completely with electrons Using the Table to Identify Valence Electrons Elements are grouped into vertical columns because they have similar properties. These are called groups or families. Groups are numbered 1-18. Group numbers can help you determine the number of valence electrons: Group 1 has 1 valence electron. Group 2 has 2 valence electrons. Groups 3–12 are transition metals and have 1 or 2 valence electrons. -

Periodic Table 1 Periodic Table
Periodic table 1 Periodic table This article is about the table used in chemistry. For other uses, see Periodic table (disambiguation). The periodic table is a tabular arrangement of the chemical elements, organized on the basis of their atomic numbers (numbers of protons in the nucleus), electron configurations , and recurring chemical properties. Elements are presented in order of increasing atomic number, which is typically listed with the chemical symbol in each box. The standard form of the table consists of a grid of elements laid out in 18 columns and 7 Standard 18-column form of the periodic table. For the color legend, see section Layout, rows, with a double row of elements under the larger table. below that. The table can also be deconstructed into four rectangular blocks: the s-block to the left, the p-block to the right, the d-block in the middle, and the f-block below that. The rows of the table are called periods; the columns are called groups, with some of these having names such as halogens or noble gases. Since, by definition, a periodic table incorporates recurring trends, any such table can be used to derive relationships between the properties of the elements and predict the properties of new, yet to be discovered or synthesized, elements. As a result, a periodic table—whether in the standard form or some other variant—provides a useful framework for analyzing chemical behavior, and such tables are widely used in chemistry and other sciences. Although precursors exist, Dmitri Mendeleev is generally credited with the publication, in 1869, of the first widely recognized periodic table. -

Recent Developments in Chalcogen Chemistry
RECENT DEVELOPMENTS IN CHALCOGEN CHEMISTRY Tristram Chivers Department of Chemistry, University of Calgary, Calgary, Alberta, Canada WHERE IS CALGARY? Lecture 1: Background / Introduction Outline • Chalcogens (O, S, Se, Te, Po) • Elemental Forms: Allotropes • Uses • Trends in Atomic Properties • Spin-active Nuclei; NMR Spectra • Halides as Reagents • Cation Formation and Stabilisation • Anions: Structures • Solutions of Chalcogens in Ionic Liquids • Oxides and Imides: Multiple Bonding 3 Elemental Forms: Sulfur Allotropes Sulfur S6 S7 S8 S10 S12 S20 4 Elemental Forms: Selenium and Tellurium Allotropes Selenium • Grey form - thermodynamically stable: helical structure cf. plastic sulfur. R. Keller, et al., Phys. Rev. B. 1977, 4404. • Red form - cyclic Cyclo-Se8 (cyclo-Se7 and -Se6 also known). Tellurium • Silvery-white, metallic lustre; helical structure, cf. grey Se. • Cyclic allotropes only known entrapped in solid-state structures e.g. Ru(Ten)Cl3 (n = 6, 8, 9) M. Ruck, Chem. Eur. J. 2011, 17, 6382 5 Uses – Sulfur Sulfur : Occurs naturally in underground deposits. • Recovered by Frasch process (superheated water). • H2S in sour gas (> 70%): Recovered by Klaus process: Klaus Process: 2 H S + SO 3/8 S + 2 H O 2 2 8 2 • Primary industrial use (70 %): H2SO4 in phosphate fertilizers 6 Uses – Selenium and Tellurium Selenium and Tellurium : Recovered during the refining of copper sulfide ores Selenium: • Photoreceptive properties – used in photocopiers (As2Se3) • Imparts red color in glasses Tellurium: • As an alloy with Cu, Fe, Pb and to harden -

• What I Hope to Convey to You Is That Rare Earths Are Uniquely Required for High Performance Magnets
• What I hope to convey to you is that rare earths are uniquely required for high performance magnets. • First a quick introduction to Arnold – the company I’ve worked for since 1992. • Arnold’s history in magnetics and magnetic materials extends back to 1895 and has included almost every commercially supplied permanent and soft magnetic product. • Today Arnold is focused on: SmCo, Alnico and bonded permanent magnets; precision thin metals – both magnetic and non-magnetic; magnetic assemblies for motors, magnetic levitation, sensing and separation technologies; and most recently we have responded to customer requests to develop and supply ultra-high performance permanent magnet motors for select applications. • Let’s start by answering this question: What makes rare earth elements so special? • The rare earth elements consist of the 15 lanthanide elements (lanthanum to lutetium) plus yttrium and scandium. • Yttrium and scandium are directly above lanthanum in the periodic table and have chemical properties that are very similar to the lanthanides – that is why they are usually included with them. • Note cesium and barium precede lanthanum in row 6 of the periodic table and that hafnium, element number 72, continues row 6 right after lutetium, the lanthanide with the highest atomic number, 71. • Note too, row 4 of the table which contains the transition elements including iron, cobalt and nickel. We’ll be making some comparison between the two groups of elements in later slides. • Rare earth elements (REEs) have considerable chemical similarities thus making them difficult to separate from each other. • That is one reason they were late in being discovered, isolated and incorporated in alloys and compounds. -

Ionic Interactions in Lanthanide Halides
Ionic Interactions in Lanthanide Halides Z. Akdeniz, Z. Çiçek, and M. P. Tosia Department of Physics, University of Istanbul, Istanbul, Turkey and Abdus Salam International Centre for Theoretical Physics, 1-34014 Trieste, Italy a INFM and Classe di Scienze, Scuola Normale Superiore, 1-56126 Pisa, Italy Reprint requests to Prof. M. P. T.; Fax: +39-050-563513; E-mail: [email protected] Z. Naturforsch. 55 a, 861-866 (2000); received September 27, 2000 We determine a model of the ionic interactions in RX3 compounds (where R is a metal in the rare-earth series from La to Lu and X = CI, Br or I) by an analysis of data on the static and dynamic structure of their molecular monomers. The potential energy function that we adopt is patterned after earlier work on Aluminium trichloride [Z. Akdeniz and M. P. Tosi, Z. Naturforsch. 54a, 180 (1999)], but includes as an essential element the electric polarizability of the trivalent metal ion to account for a pyramidal shape of RX3 molecules. From data referring mostly to trihalides of elements at the ends and in the middle of the rare-earth series (/. e. LaX3, GdX3 and LuX3), we propose systematic variations for the effective valence, ionic radius and electric polarizability of the metal ions across the series. As a first application of our results we predict the structure of the Dy2Cl6 and Dy2Br6 molecular dimers and demonstrate by comparison with electron diffraction data that lanthanide-ion polarizability plays a quantitative role also in this state of tetrahedral-like coordination. Key words: Ionic Clusters; Molten Salts. -

Honors Chemistry ___/___/___
Name ____________________ Honors Chemistry ___/___/___ Chapter 3 Practice Test Part I: For each of the following, write the symbol of the element that best fits the description given. You may use an element more than once; you may even use it more than twice. (2 point each) 1. Li The alkali metal with the greatest ionization energy. 2. F The halogen with the greatest electronegativity. 3. Rn The noble gas with the greatest atomic radius. 4. Br The fourth period element with the highest electron affinity. 5. Pm The synthetic rare-earth element with the smallest atomic number. 6. Be The alkaline-earth element with the greatest second ionization energy. 7. Sb The fifth period metalloid with the largest atomic radius. 8. He The element on the periodic table with the smallest atomic radius. 9. Ar The third period non-metal with the highest ionization energy. 10. C The second period non-metal with the fewest valence electrons. 11. U The naturally occurring element with the greatest atomic mass. 12. Ne The second period element with the greatest ionization energy. 13. He The element with the greatest ionization energy. 14. Se The chalcogen non-metal with the greatest number of protons. 15. Al The third period metal with a 3+ oxidation number. 16. Fr The element with the lowest ionization energy. 17. Be The alkaline-earth element with the smallest atomic radius. 18. F The element with the greatest electronegativity. 19. At The sixth period element with the greatest electronegativity. 20. B The metalloid with the fewest valence electrons. -

Conceptual Progress for Explaining and Predicting Self-Organization on Anodized Aluminum Surfaces
nanomaterials Review Conceptual Progress for Explaining and Predicting Self-Organization on Anodized Aluminum Surfaces Mikhail Pashchanka Department of Chemistry, Eduard-Zintl-Institute, Technical University of Darmstadt, Alarich-Weiss-Straße 12, 64287 Darmstadt, Germany; [email protected] Abstract: Over the past few years, researchers have made numerous breakthroughs in the field of aluminum anodizing and faced the problem of the lack of adequate theoretical models for the interpretation of some new experimental findings. For instance, spontaneously formed anodic alumina nanofibers and petal-like patterns, flower-like structures observed under AC anodizing conditions, and hierarchical pores whose diameters range from several nanometers to sub-millimeters could be explained neither by the classical field-assisted dissolution theory nor by the plastic flow model. In addition, difficulties arose in explaining the basic indicators of porous film growth, such as the nonlinear current–voltage characteristics of electrochemical cells or the evolution of hexagonal pore patterns at the early stages of anodizing experiments. Such a conceptual crisis resulted in new multidisciplinary investigations and the development of novel theoretical models, whose evolution is discussed at length in this review work. The particular focus of this paper is on the recently developed electroconvection-based theories that allowed making truly remarkable advances in understanding the porous anodic alumina formation process in the last 15 years. Some explanation of the synergy between electrode reactions and transport processes leading to self-organization is provided. Finally, future prospects for the synthesis of novel anodic architectures are discussed. Citation: Pashchanka, M. Conceptual Progress for Explaining Keywords: porous anodic alumina (PAA); chaos and self-organization theory; electroconvection; and Predicting Self-Organization on colloidal gel model; anion exchange; DLVO theory; fluid mechanics; surface chemistry; surface Anodized Aluminum Surfaces.