Emergence of Californium As the Second Transitional Element in the Actinide Series
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Table 2.Iii.1. Fissionable Isotopes1
FISSIONABLE ISOTOPES Charles P. Blair Last revised: 2012 “While several isotopes are theoretically fissionable, RANNSAD defines fissionable isotopes as either uranium-233 or 235; plutonium 238, 239, 240, 241, or 242, or Americium-241. See, Ackerman, Asal, Bale, Blair and Rethemeyer, Anatomizing Radiological and Nuclear Non-State Adversaries: Identifying the Adversary, p. 99-101, footnote #10, TABLE 2.III.1. FISSIONABLE ISOTOPES1 Isotope Availability Possible Fission Bare Critical Weapon-types mass2 Uranium-233 MEDIUM: DOE reportedly stores Gun-type or implosion-type 15 kg more than one metric ton of U- 233.3 Uranium-235 HIGH: As of 2007, 1700 metric Gun-type or implosion-type 50 kg tons of HEU existed globally, in both civilian and military stocks.4 Plutonium- HIGH: A separated global stock of Implosion 10 kg 238 plutonium, both civilian and military, of over 500 tons.5 Implosion 10 kg Plutonium- Produced in military and civilian 239 reactor fuels. Typically, reactor Plutonium- grade plutonium (RGP) consists Implosion 40 kg 240 of roughly 60 percent plutonium- Plutonium- 239, 25 percent plutonium-240, Implosion 10-13 kg nine percent plutonium-241, five 241 percent plutonium-242 and one Plutonium- percent plutonium-2386 (these Implosion 89 -100 kg 242 percentages are influenced by how long the fuel is irradiated in the reactor).7 1 This table is drawn, in part, from Charles P. Blair, “Jihadists and Nuclear Weapons,” in Gary A. Ackerman and Jeremy Tamsett, ed., Jihadists and Weapons of Mass Destruction: A Growing Threat (New York: Taylor and Francis, 2009), pp. 196-197. See also, David Albright N 2 “Bare critical mass” refers to the absence of an initiator or a reflector. -

The Development of the Periodic Table and Its Consequences Citation: J
Firenze University Press www.fupress.com/substantia The Development of the Periodic Table and its Consequences Citation: J. Emsley (2019) The Devel- opment of the Periodic Table and its Consequences. Substantia 3(2) Suppl. 5: 15-27. doi: 10.13128/Substantia-297 John Emsley Copyright: © 2019 J. Emsley. This is Alameda Lodge, 23a Alameda Road, Ampthill, MK45 2LA, UK an open access, peer-reviewed article E-mail: [email protected] published by Firenze University Press (http://www.fupress.com/substantia) and distributed under the terms of the Abstract. Chemistry is fortunate among the sciences in having an icon that is instant- Creative Commons Attribution License, ly recognisable around the world: the periodic table. The United Nations has deemed which permits unrestricted use, distri- 2019 to be the International Year of the Periodic Table, in commemoration of the 150th bution, and reproduction in any medi- anniversary of the first paper in which it appeared. That had been written by a Russian um, provided the original author and chemist, Dmitri Mendeleev, and was published in May 1869. Since then, there have source are credited. been many versions of the table, but one format has come to be the most widely used Data Availability Statement: All rel- and is to be seen everywhere. The route to this preferred form of the table makes an evant data are within the paper and its interesting story. Supporting Information files. Keywords. Periodic table, Mendeleev, Newlands, Deming, Seaborg. Competing Interests: The Author(s) declare(s) no conflict of interest. INTRODUCTION There are hundreds of periodic tables but the one that is widely repro- duced has the approval of the International Union of Pure and Applied Chemistry (IUPAC) and is shown in Fig.1. -

Actinide Ground-State Properties-Theoretical Predictions
Actinide Ground-State Properties Theoretical predictions John M. Wills and Olle Eriksson electron-electron correlations—the electronic energy of the ground state of or nearly fifty years, the actinides interactions among the 5f electrons and solids, molecules, and atoms as a func- defied the efforts of solid-state between them and other electrons—are tional of electron density. The DFT Ftheorists to understand their expected to affect the bonding. prescription has had such a profound properties. These metals are among Low-symmetry crystal structures, impact on basic research in both the most complex of the long-lived relativistic effects, and electron- chemistry and solid-state physics that elements, and in the solid state, they electron correlations are very difficult Walter Kohn, its main inventor, was display some of the most unusual to treat in traditional electronic- one of the recipients of the 1998 behaviors of any series in the periodic structure calculations of metals and, Nobel Prize in Chemistry. table. Very low melting temperatures, until the last decade, were outside the In general, it is not possible to apply large anisotropic thermal-expansion realm of computational ability. And DFT without some approximation. coefficients, very low symmetry crystal yet, it is essential to treat these effects But many man-years of intense research structures, many solid-to-solid phase properly in order to understand the have yielded reliable approximate transitions—the list is daunting. Where physics of the actinides. Electron- expressions for the total energy in does one begin to put together an electron correlations are important in which all terms, except for a single- understanding of these elements? determining the degree to which 5f particle kinetic-energy term, can be In the last 10 years, together with electrons are localized at lattice sites. -

Health Physics Education Reference Book
HEALTH PHYSICS EDUCATION REFERENCE BOOK 2010 - 2011 Health Physics Society Academic Education Committee Updated June 2010 1. Bloomsburg University Pennsylvania BS 2. Clemson University South Carolina MS PhD 3. Colorado State University Colorado MS PhD 4. Duke University North Carolina MS PhD 5. Francis Marion University South Carolina BS 6. Idaho State University Idaho AA BS MS PhD 7. Illinois Institute of Technology Illinois MS 8. Linn State Technical College Missouri AA 9. Louisiana State University Louisiana MS PhD 10. Ohio State University Ohio MS PhD 11. Oregon State University Oregon BS MS PhD 12. Purdue University Indiana BS MS PhD 13. Rensselaer Polytechnic Institute New York BS MS PhD 14. San Diego State University California MS 15. Texas A&M University Texas BS MS PhD 16. Texas State Technical College Texas AA 17. Thomas Edison State College AS BS 18. University of Cincinnati Ohio MS PhD 19. University of Florida Florida BS MS PhD 20. University of Massachusetts Lowell Massachusetts BS MS PhD 21. University of Michigan Michigan BS MS PhD 22. University of Missouri-Columbia Missouri MS PhD 23. University of Nevada Las Vegas Nevada BS MS 24. University of Tennessee Tennessee BS MS PhD 25. Vanderbilt University Tennessee MS PhD 26. Virginia Commonwealth University Degree Programs Recognized by the Accreditation Board for Engineering and Technology (ABET) in Health Physics under ABET’s Applied Science Accreditation Commission (ASAC) Bloomsburg University Health Physics (BS) (2006) Clemson University Environmental Health Physics (MS) (2005) Colorado State University Health Physics (MS) (2007) Idaho State University Health Physics (BS) (2003) Idaho State University Health Physics (MS) (2003) Oregon State University Radiation (2004) University of Nevada Las Vegas Health Physics (MS) (2003) Degree Programs Recognized by the Accreditation Board for Engineering and Technology (ABET) in Radiological Engineering under ABET’s Engineering Accreditation Commission (EAC) Texas A&M University Radiological Health Engineering (BS) (1987) 1. -
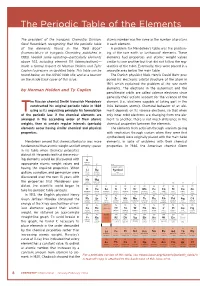
The Periodic Table of the Elements
The Periodic Table of the Elements The president of the Inorganic Chemistry Division, atomic number was the same as the number of protons Gerd Rosenblatt, recognizing that the periodic table in each element. of the elements found in the “Red Book” A problem for Mendeleev’s table was the position- (Nomenclature of Inorganic Chemistry, published in ing of the rare earth or lanthanoid* elements. These 1985) needed some updating—particularly elements elements had properties and atomic weight values above 103, including element 110 (darmstadtium)— similar to one another but that did not follow the reg- made a formal request to Norman Holden and Tyler ularities of the table. Eventually, they were placed in a Coplen to prepare an updated table. This table can be separate area below the main table. found below, on the IUPAC Web site, and as a tear-off The Danish physicist Niels Henrik David Bohr pro- on the inside back cover of this issue. posed his electronic orbital structure of the atom in 1921, which explained the problem of the rare earth by Norman Holden and Ty Coplen elements. The electrons in the outermost and the penultimate orbits are called valence electrons since generally their actions account for the valence of the he Russian chemist Dmitri Ivanovich Mendeleev element (i.e., electrons capable of taking part in the constructed his original periodic table in 1869 links between atoms). Chemical behavior of an ele- Tusing as its organizing principle his formulation ment depends on its valence electrons, so that when of the periodic law: if the chemical elements are only inner orbit electrons are changing from one ele- arranged in the ascending order of their atomic ment to another, there is not much difference in the weights, then at certain regular intervals (periods) chemical properties between the elements. -

ELECTRONEGATIVITY D Qkq F=
ELECTRONEGATIVITY The electronegativity of an atom is the attracting power that the nucleus has for it’s own outer electrons and those of it’s neighbours i.e. how badly it wants electrons. An atom’s electronegativity is determined by Coulomb’s Law, which states, “ the size of the force is proportional to the size of the charges and inversely proportional to the square of the distance between them”. In symbols it is represented as: kq q F = 1 2 d 2 where: F = force (N) k = constant (dependent on the medium through which the force is acting) e.g. air q1 = charge on an electron (C) q2 = core charge (C) = no. of protons an outer electron sees = no. of protons – no. of inner shell electrons = main Group Number d = distance of electron from the nucleus (m) Examples of how to calculate the core charge: Sodium – Atomic number 11 Electron configuration 2.8.1 Core charge = 11 (no. of p+s) – 10 (no. of inner e-s) +1 (Group i) Chlorine – Atomic number 17 Electron configuration 2.8.7 Core charge = 17 (no. of p+s) – 10 (no. of inner e-s) +7 (Group vii) J:\Sciclunm\Resources\Year 11 Chemistry\Semester1\Notes\Atoms & The Periodic Table\Electronegativity.doc Neon holds onto its own electrons with a core charge of +8, but it can’t hold any more electrons in that shell. If it was to form bonds, the electron must go into the next shell where the core charge is zero. Group viii elements do not form any compounds under normal conditions and are therefore given no electronegativity values. -

Periodic Table of the Elements Notes
Periodic Table of the Elements Notes Arrangement of the known elements based on atomic number and chemical and physical properties. Divided into three basic categories: Metals (left side of the table) Nonmetals (right side of the table) Metalloids (touching the zig zag line) Basic Organization by: Atomic structure Atomic number Chemical and Physical Properties Uses of the Periodic Table Useful in predicting: chemical behavior of the elements trends properties of the elements Atomic Structure Review: Atoms are made of protons, electrons, and neutrons. Elements are atoms of only one type. Elements are identified by the atomic number (# of protons in nucleus). Energy Levels Review: Electrons are arranged in a region around the nucleus called an electron cloud. Energy levels are located within the cloud. At least 1 energy level and as many as 7 energy levels exist in atoms Energy Levels & Valence Electrons Energy levels hold a specific amount of electrons: 1st level = up to 2 2nd level = up to 8 3rd level = up to 8 (first 18 elements only) The electrons in the outermost level are called valence electrons. Determine reactivity - how elements will react with others to form compounds Outermost level does not usually fill completely with electrons Using the Table to Identify Valence Electrons Elements are grouped into vertical columns because they have similar properties. These are called groups or families. Groups are numbered 1-18. Group numbers can help you determine the number of valence electrons: Group 1 has 1 valence electron. Group 2 has 2 valence electrons. Groups 3–12 are transition metals and have 1 or 2 valence electrons. -

Periodic Table 1 Periodic Table
Periodic table 1 Periodic table This article is about the table used in chemistry. For other uses, see Periodic table (disambiguation). The periodic table is a tabular arrangement of the chemical elements, organized on the basis of their atomic numbers (numbers of protons in the nucleus), electron configurations , and recurring chemical properties. Elements are presented in order of increasing atomic number, which is typically listed with the chemical symbol in each box. The standard form of the table consists of a grid of elements laid out in 18 columns and 7 Standard 18-column form of the periodic table. For the color legend, see section Layout, rows, with a double row of elements under the larger table. below that. The table can also be deconstructed into four rectangular blocks: the s-block to the left, the p-block to the right, the d-block in the middle, and the f-block below that. The rows of the table are called periods; the columns are called groups, with some of these having names such as halogens or noble gases. Since, by definition, a periodic table incorporates recurring trends, any such table can be used to derive relationships between the properties of the elements and predict the properties of new, yet to be discovered or synthesized, elements. As a result, a periodic table—whether in the standard form or some other variant—provides a useful framework for analyzing chemical behavior, and such tables are widely used in chemistry and other sciences. Although precursors exist, Dmitri Mendeleev is generally credited with the publication, in 1869, of the first widely recognized periodic table. -
The Impact of Partitioning and Transmutation on the Risk Assesment of a Spent Nuclear Fuel
h/e/-se SE0608415 The Impact of Partitioning and Transmutation on the Risk Assesment of a Spent Nuclear Fuel Naima Amrani and Ahmed Boucenna UFAS University Physics Department Faculty of sciences Setif 19000 ALGERIA I. INTRODUCTION Nuclear power produces steadily a mass of spent fuel which contains a part from the short lived fission products, a significant amount of actinides and fission products with height toxicity and long half-lives. These nuclides constitute the long-term radiotoxic inventory which remains as a hazard far beyond human perception. The reprocessing recycles most of the major actinides (Uranium and Plutonium), while the Minor Actinides (MA) (mainly Neptunium: Np, Americium: Am and Curium: Cm) with half lives up to 2 million years remain with the fission products which are vitrified before being buried in deep repositories. Partitioning of the minor actinides and some of the fission products is an efficient method to reduce the long term radiotoxicity of the residual waste components with a factor proportional to the separation yield. The improved minor actinide nuclides would be recycled into a fuel cycle activities and returned to the reactor inventory of fissile and fertile material for transmutation to short lived isotopes. Progressively the MAs and some long-lived fission product (LLFP) could be burn up out. This option would reduce the long term contamination hazard in the high-level waste and shorten the time interval necessary to keep the actinides containing wastes confined in a deep geologic repository. Partitioning and Transmutation (P&T) is, in principal, capable of reducing the radiotoxicity period, while a number of practical difficult remain to be surmounted. -

Monte Carlo Modelling of Th-Pb Fuel Assembly with Californium Neutron Source 89
NUKLEONIKA 2018;63(3):8791 doi: 10.2478/nuka-2018-0011 ORIGINAL PAPER © 2018 M. Oettingen and P. Stanisz. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 3.0 License (CC BY-NC-ND 3.0). Monte Carlo modelling of Th-Pb fuel assembly Mikołaj Oettingen, with californium neutron source Przemysław Stanisz Abstract. This paper describes the methodology developed for the numerical reconstruction and modelling of the thorium-lead (Th-Pb) assembly available at the Department of Nuclear Energy, Faculty of Energy and Fuels, AGH University, Krakow, Poland. This numerical study is the fi rst step towards integral irradiation experiments in the Th-Pb environment. The continuous-energy Monte Carlo burnup (MCB) code available on supercomputer Prometheus of ACK Cyfronet AGH was applied for numerical modelling. The assembly consists of a hexagonal array of ThO2 fuel rods and metallic Pb rods. The design allows for different arrangements of the rods for var- ious types of irradiations and experimental measurements. The intensity of the fresh neutron source intended for integral experiments is about 108 n/s, which corresponds to the mass of about 43 g 252Cf. The source was modelled in the form of Cf2O3-Pd cermet wire embedded in two stainless steel capsules. Keywords: californium • lead • Monte Carlo • MCB • thorium Introduction Current light-water nuclear reactors operate on the enriched uranium fuel. The main fi ssile isotope in such fuel is 235U; however, in a series of transmuta- tions and decays, fi ssile isotopes of plutonium (i.e. 239Pu and 241Pu) are formed. In general, about 70% of fi ssions during a reactor cycle in commercial nuclear reactors occur on 235U, whereas about 30% of fi ssions occur on 239Pu and 241Pu. -

Genius of the Periodic Table
GENIUS OF THE PERIODIC TABLE "Isn't it the work of a genius'. " exclaimed Academician V.I. Spitsyn, USSR, a member of the Scientific Advisory Committee when talking to an Agency audience in January. His listeners shared his enthusiasm. Academician Spitsyn was referring to the to the first formulation a hundred years ago by Professor Dmitry I. Mendeleyev of the Periodic Law of Elements. In conditions of enormous difficulty, considering the lack of data on atomic weights of elements, Mendeleyev created in less than two years work at St. Petersburg University, a system of chemical elements that is, in general, still being used. His law became a powerful instrument for further development of chemistry and physics. He was able immediately to correct the atomic weight numbers of some elements, including uranium, whose atomic weight he found to be double that given at the time. Two years later Mendeleyev went so far as to give a detailed description of physical or chemical properties of some elements which were as yet undiscovered. Time gave striking proof of his predictions and his periodic law. Mendeleyev published his conclusions in the first place by sending, early in March 186 9, a leaflet to many Russian and foreign scientists. It gave his system of elements based on their atomic weights and chemical resemblance. On the 18th March that year his paper on the subject was read at the meeting of the Russian Chemical Society, and two months later the Society's Journal published his article entitled "The correlation between properties of elements and their atomic weight". -

Plutonium Incorporation in Phosphate and Titanate Ceramics for Minor Actinide Containment
Journal of Nuclear Materials 352 (2006) 233–240 www.elsevier.com/locate/jnucmat Plutonium incorporation in phosphate and titanate ceramics for minor actinide containment X. Deschanels a,*, V. Picot a, B. Glorieux b, F. Jorion a, S. Peuget a, D. Roudil a, C. Je´gou a, V. Broudic a, J.N. Cachia a, T. Advocat a, C. Den Auwer a, C. Fillet a, J.P. Coutures b, C. Hennig c, A. Scheinost c a DIEC/LMPA, CEA/DEN Valrho Marcoule, BP 17171, 30207 Bagnols-sur-Ce`ze cedex, France b PROMES-CNRS Tecnosud, Rambla de la Thermodynamique, 66100 Perpignan, France c ROBL-CRG, ESRF, BP 220, 38043 Grenoble, France Abstract Two ceramics, zirconolite and a monazite–brabantite solid solution (MBss) were studied for the immobilization of minor actinides (Np, Am, Cm) produced by reprocessing spent fuel. Monoclinic zirconolite (CaZrTi2O7) is a fluorite deriv- ative structure and is the primary actinide host phase in Synroc (a titanate composite). Monazite (LnPO4, where Ln = La, Ce, Nd, Gd, etc.) is a monoclinic orthophosphate containing trivalent cations, and brabantite (Ca0.5An0.5PO4) is an iso- structural monazite compound containing tetravalent cations (An = Th and U). The nominal composition of the ceramics studied in this work is (Ca0.87Pu0.13)Zr(Al0. 26Ti1.74)O7 for zirconolite and (Ca0.09Pu0.09La0.73Th0.09)PO4 for the monazite– brabantite solid solution. These formulas correspond to 10 wt% PuO2 loading in each material. XANES spectroscopy showed that the plutonium is tetravalent in zirconolite and trivalent in MBss. Thorium, another tetravalent cation, can be incorporated at 10 wt% ThO2 in MBss.