Draft Genome Sequence of the Agarase-Producing Sphingomonas Sp
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Characterization of the Aerobic Anoxygenic Phototrophic Bacterium Sphingomonas Sp
microorganisms Article Characterization of the Aerobic Anoxygenic Phototrophic Bacterium Sphingomonas sp. AAP5 Karel Kopejtka 1 , Yonghui Zeng 1,2, David Kaftan 1,3 , Vadim Selyanin 1, Zdenko Gardian 3,4 , Jürgen Tomasch 5,† , Ruben Sommaruga 6 and Michal Koblížek 1,* 1 Centre Algatech, Institute of Microbiology, Czech Academy of Sciences, 379 81 Tˇreboˇn,Czech Republic; [email protected] (K.K.); [email protected] (Y.Z.); [email protected] (D.K.); [email protected] (V.S.) 2 Department of Plant and Environmental Sciences, University of Copenhagen, Thorvaldsensvej 40, 1871 Frederiksberg C, Denmark 3 Faculty of Science, University of South Bohemia, 370 05 Ceskˇ é Budˇejovice,Czech Republic; [email protected] 4 Institute of Parasitology, Biology Centre, Czech Academy of Sciences, 370 05 Ceskˇ é Budˇejovice,Czech Republic 5 Research Group Microbial Communication, Technical University of Braunschweig, 38106 Braunschweig, Germany; [email protected] 6 Laboratory of Aquatic Photobiology and Plankton Ecology, Department of Ecology, University of Innsbruck, 6020 Innsbruck, Austria; [email protected] * Correspondence: [email protected] † Present Address: Department of Molecular Bacteriology, Helmholtz-Centre for Infection Research, 38106 Braunschweig, Germany. Abstract: An aerobic, yellow-pigmented, bacteriochlorophyll a-producing strain, designated AAP5 Citation: Kopejtka, K.; Zeng, Y.; (=DSM 111157=CCUG 74776), was isolated from the alpine lake Gossenköllesee located in the Ty- Kaftan, D.; Selyanin, V.; Gardian, Z.; rolean Alps, Austria. Here, we report its description and polyphasic characterization. Phylogenetic Tomasch, J.; Sommaruga, R.; Koblížek, analysis of the 16S rRNA gene showed that strain AAP5 belongs to the bacterial genus Sphingomonas M. Characterization of the Aerobic and has the highest pairwise 16S rRNA gene sequence similarity with Sphingomonas glacialis (98.3%), Anoxygenic Phototrophic Bacterium Sphingomonas psychrolutea (96.8%), and Sphingomonas melonis (96.5%). -

Proposal of Sphingomonadaceae Fam. Nov., Consisting of Sphingomonas Yabuuchi Et Al. 1990, Erythrobacter Shiba and Shimidu 1982, Erythromicrobium Yurkov Et Al
Microbiol. Immunol., 44(7), 563-575, 2000 Proposal of Sphingomonadaceae Fam. Nov., Consisting of Sphingomonas Yabuuchi et al. 1990, Erythrobacter Shiba and Shimidu 1982, Erythromicrobium Yurkov et al. 1994, Porphyrobacter Fuerst et al. 1993, Zymomonas Kluyver and van Niel 1936, and Sandaracinobacter Yurkov et al. 1997, with the Type Genus Sphingomonas Yabuuchi et al. 1990 Yoshimasa Kosako*°', Eiko Yabuuchi2, Takashi Naka3,4, Nagatoshi Fujiwara3, and Kazuo Kobayashi3 'JapanCollection of Microorganis ms,RIKEN (Institute of Physical and ChemicalResearch), Wako, Saitama 351-0198, Japan, 2Departmentof Microbiologyand Immunology , AichiMedical University, Aichi 480-1101, Japan, 'Departmentof Host Defense,Osaka City University, Graduate School of Medicine,Osaka, Osaka 545-8585, Japan, and Instituteof SkinSciences, ClubCosmetics Co., Ltd., Osaka,Osaka 550-0005, Japan ReceivedJanuary 25, 2000; in revisedform, April 11, 2000. Accepted April 14, 2000 Abstract:Based on the results of phylogeneticanalysis of the 16SrDNA sequences and the presence of N- 2'-hydroxymyristoyldihydrosphingosine 1-glucuronic acid (SGL-1)and 2-hydroxymyristicacid (non- hydroxymyristicacid in Zymomonas)in cellular lipids,a new family,Sphingomonadaceae, for Group 4 of the alpha-subclassof the classProteobacteria is hereinproposed and a descriptionof the familyis given.The familyconsists of six genera, Sphingomonas,Erythrobacter, Erythromicrobium, Porphyrobacter, Sandara- cinobacterand Zymomonas.Thus, all the validlypublished and currently known genera in Group 4 of the alpha-subclassof -

Polyamine Profiles of Some Members of the Alpha Subclass of the Class Proteobacteria: Polyamine Analysis of Twenty Recently Described Genera
Microbiol. Cult. Coll. June 2003. p. 13 ─ 21 Vol. 19, No. 1 Polyamine Profiles of Some Members of the Alpha Subclass of the Class Proteobacteria: Polyamine Analysis of Twenty Recently Described Genera Koei Hamana1)*,Azusa Sakamoto1),Satomi Tachiyanagi1), Eri Terauchi1)and Mariko Takeuchi2) 1)Department of Laboratory Sciences, School of Health Sciences, Faculty of Medicine, Gunma University, 39 ─ 15 Showa-machi 3 ─ chome, Maebashi, Gunma 371 ─ 8514, Japan 2)Institute for Fermentation, Osaka, 17 ─ 85, Juso-honmachi 2 ─ chome, Yodogawa-ku, Osaka, 532 ─ 8686, Japan Cellular polyamines of 41 newly validated or reclassified alpha proteobacteria belonging to 20 genera were analyzed by HPLC. Acetic acid bacteria belonging to the new genus Asaia and the genera Gluconobacter, Gluconacetobacter, Acetobacter and Acidomonas of the alpha ─ 1 sub- group ubiquitously contained spermidine as the major polyamine. Aerobic bacteriochlorophyll a ─ containing Acidisphaera, Craurococcus and Paracraurococcus(alpha ─ 1)and Roseibium (alpha-2)contained spermidine and lacked homospermidine. New Rhizobium species, including some species transferred from the genera Agrobacterium and Allorhizobium, and new Sinorhizobium and Mesorhizobium species of the alpha ─ 2 subgroup contained homospermidine as a major polyamine. Homospermidine was the major polyamine in the genera Oligotropha, Carbophilus, Zavarzinia, Blastobacter, Starkeya and Rhodoblastus of the alpha ─ 2 subgroup. Rhodobaca bogoriensis of the alpha ─ 3 subgroup contained spermidine. Within the alpha ─ 4 sub- group, the genus Sphingomonas has been divided into four clusters, and species of the emended Sphingomonas(cluster I)contained homospermidine whereas those of the three newly described genera Sphingobium, Novosphingobium and Sphingopyxis(corresponding to clusters II, III and IV of the former Sphingomonas)ubiquitously contained spermidine. -

Evolutionary Genomics of an Ancient Prophage of the Order Sphingomonadales
GBE Evolutionary Genomics of an Ancient Prophage of the Order Sphingomonadales Vandana Viswanathan1,2, Anushree Narjala1, Aravind Ravichandran1, Suvratha Jayaprasad1,and Shivakumara Siddaramappa1,* 1Institute of Bioinformatics and Applied Biotechnology, Biotech Park, Electronic City, Bengaluru, Karnataka, India 2Manipal University, Manipal, Karnataka, India *Corresponding author: E-mail: [email protected]. Accepted: February 10, 2017 Data deposition: Genome sequences were downloaded from GenBank, and their accession numbers are provided in table 1. Abstract The order Sphingomonadales, containing the families Erythrobacteraceae and Sphingomonadaceae, is a relatively less well-studied phylogenetic branch within the class Alphaproteobacteria. Prophage elements are present in most bacterial genomes and are important determinants of adaptive evolution. An “intact” prophage was predicted within the genome of Sphingomonas hengshuiensis strain WHSC-8 and was designated Prophage IWHSC-8. Loci homologous to the region containing the first 22 open reading frames (ORFs) of Prophage IWHSC-8 were discovered among the genomes of numerous Sphingomonadales.In17genomes, the homologous loci were co-located with an ORF encoding a putative superoxide dismutase. Several other lines of molecular evidence implied that these homologous loci represent an ancient temperate bacteriophage integration, and this horizontal transfer event pre-dated niche-based speciation within the order Sphingomonadales. The “stabilization” of prophages in the genomes of their hosts is an indicator of “fitness” conferred by these elements and natural selection. Among the various ORFs predicted within the conserved prophages, an ORF encoding a putative proline-rich outer membrane protein A was consistently present among the genomes of many Sphingomonadales. Furthermore, the conserved prophages in six Sphingomonas sp. contained an ORF encoding a putative spermidine synthase. -

A Primary Assessment of the Endophytic Bacterial Community in a Xerophilous Moss (Grimmia Montana) Using Molecular Method and Cultivated Isolates
Brazilian Journal of Microbiology 45, 1, 163-173 (2014) Copyright © 2014, Sociedade Brasileira de Microbiologia ISSN 1678-4405 www.sbmicrobiologia.org.br Research Paper A primary assessment of the endophytic bacterial community in a xerophilous moss (Grimmia montana) using molecular method and cultivated isolates Xiao Lei Liu, Su Lin Liu, Min Liu, Bi He Kong, Lei Liu, Yan Hong Li College of Life Science, Capital Normal University, Haidian District, Beijing, China. Submitted: December 27, 2012; Approved: April 1, 2013. Abstract Investigating the endophytic bacterial community in special moss species is fundamental to under- standing the microbial-plant interactions and discovering the bacteria with stresses tolerance. Thus, the community structure of endophytic bacteria in the xerophilous moss Grimmia montana were esti- mated using a 16S rDNA library and traditional cultivation methods. In total, 212 sequences derived from the 16S rDNA library were used to assess the bacterial diversity. Sequence alignment showed that the endophytes were assigned to 54 genera in 4 phyla (Proteobacteria, Firmicutes, Actinobacteria and Cytophaga/Flexibacter/Bacteroids). Of them, the dominant phyla were Proteobacteria (45.9%) and Firmicutes (27.6%), the most abundant genera included Acinetobacter, Aeromonas, Enterobacter, Leclercia, Microvirga, Pseudomonas, Rhizobium, Planococcus, Paenisporosarcina and Planomicrobium. In addition, a total of 14 species belonging to 8 genera in 3 phyla (Proteo- bacteria, Firmicutes, Actinobacteria) were isolated, Curtobacterium, Massilia, Pseudomonas and Sphingomonas were the dominant genera. Although some of the genera isolated were inconsistent with those detected by molecular method, both of two methods proved that many different endophytic bacteria coexist in G. montana. According to the potential functional analyses of these bacteria, some species are known to have possible beneficial effects on hosts, but whether this is the case in G. -

Characterization of the Microbiome of Nipple Aspirate Fluid of Breast Cancer Survivors Received: 23 March 2016 Alfred A
www.nature.com/scientificreports OPEN Characterization of the microbiome of nipple aspirate fluid of breast cancer survivors Received: 23 March 2016 Alfred A. Chan1,*, Mina Bashir2,3,*, Magali N. Rivas1,*, Karen Duvall4,5, Peter A. Sieling1, Accepted: 31 May 2016 Thomas R. Pieber3, Parag A. Vaishampayan2, Susan M. Love5 & Delphine J. Lee1 Published: 21 June 2016 The microbiome impacts human health and disease. Until recently, human breast tissue and milk were presumed to be sterile. Here, we investigated the presence of microbes in the nipple aspirate fluid (NAF) and their potential association with breast cancer. We compared the NAF microbiome between women with a history of breast cancer (BC) and healthy control women (HC) using 16S rRNA gene amplicon sequencing. The NAF microbiome from BC and HC showed significant differences in community composition. Two Operational Taxonomic Units (OTUs) showed differences in relative abundances between NAF collected from BC and HC. In NAF collected from BC, there was relatively higher incidence of the genus Alistipes. By contrast, an unclassified genus from theSphingomonadaceae family was relatively more abundant in NAF from HC. These findings reflect the ductal source DNA since there were no differences between areolar skin samples collected from BC and HC. Furthermore, the microbes associated with BC share an enzymatic activity, Beta-Glucuronidase, which may promote breast cancer. This is the first report of bacterial DNA in human breast ductal fluid and the differences between NAF from HC and BC. Further investigation of the ductal microbiome and its potential role in breast cancer are warranted. The human microbiome is the term applied to the universe of microbes that inhabit our skin and mucosal surfaces. -

Microbial Community Structure Analysis in Acer Palmatum Bark and Isolation of Novel Bacteria IAD-21 of the Phylum Abditibacteriota (Former Candidate Division FBP)
Microbial community structure analysis in Acer palmatum bark and isolation of novel bacteria IAD-21 of the phylum Abditibacteriota (former candidate division FBP) Kazuki Kobayashi1 and Hideki Aoyagi1,2 1 Division of Life Sciences and Bioengineering, Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Ibaraki, Japan 2 Faculty of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Ibaraki, Japan ABSTRACT Background: The potential of unidentified microorganisms for academic and other applications is limitless. Plants have diverse microbial communities associated with their biomes. However, few studies have focused on the microbial community structure relevant to tree bark. Methods: In this report, the microbial community structure of bark from the broad-leaved tree Acer palmatum was analyzed. Both a culture-independent approach using polymerase chain reaction (PCR) amplification and next generation sequencing, and bacterial isolation and sequence-based identification methods were used to explore the bark sample as a source of previously uncultured microorganisms. Molecular phylogenetic analyses based on PCR-amplified 16S rDNA sequences were performed. Results: At the phylum level, Proteobacteria and Bacteroidetes were relatively abundant in the A. palmatum bark. In addition, microorganisms from the phyla Acidobacteria, Gemmatimonadetes, Verrucomicrobia, Armatimonadetes, and Submitted 2 February 2019 Abditibacteriota, which contain many uncultured microbial species, existed in the Accepted 12 September 2019 A. palmatum bark. Of the 30 genera present at relatively high abundance in the bark, Published 29 October 2019 some genera belonging to the phyla mentioned were detected. A total of 70 isolates Corresponding author could be isolated and cultured using the low-nutrient agar media DR2A and Hideki Aoyagi, PE03. -

Sphingomonas Solaris Sp. Nov., Isolated from a Solar Panel in Boston, Massachusetts
TAXONOMIC DESCRIPTION Tanner et al., Int. J. Syst. Evol. Microbiol. 2020;70:1814–1821 DOI 10.1099/ijsem.0.003977 Sphingomonas solaris sp. nov., isolated from a solar panel in Boston, Massachusetts Kristie Tanner1,2,3,4, Christopher P. Mancuso5,6, Juli Peretó1,4,7, Ahmad S. Khalil3,5,6, Cristina Vilanova1 and Javier Pascual1,* Abstract Solar panel surfaces, although subjected to a range of extreme environmental conditions, are inhabited by a diverse microbial community adapted to solar radiation, desiccation and temperature fluctuations. This is the first time a new bacterial species has been isolated from this environment. Strain R4DWNT belongs to the genus Sphingomonas and was isolated from a solar panel surface in Boston, MA, USA. Strain R4DWNT is a Gram- negative, non-motile and rod- shaped bacteria that tested posi- tive for oxidase and catalase and forms round-shaped, shiny and orange- coloured colonies. It is mesophilic, neutrophilic and non- halophilic, and presents a more stenotrophic metabolism than its closest neighbours. The major fatty acids in this strain are C18:1ω7c/C18:1ω6c, C16:1ω7c/C16:1ω6c, C14:0 2OH and C16:0. Comparison of 16S rRNA gene sequences revealed that the closest type strains to R4DWNT are Sphingomonas fennica, Sphingomonas formosensis, Sphingomonas prati, Sphingomonas montana and Sphingomonas oleivorans with 96.3, 96.1, 96.0, 95.9 and 95.7 % pairwise similarity, respectively. The genomic G+C content of R4DWNT is 67.9 mol%. Based on these characteristics, strain R4DWNT represents a novel species of the genus Sphingomonas for which the name Sphingomonas solaris sp. nov. is proposed with the type strain R4DWNT (=CECT 9811T=LMG 31344T). -

Polycyclic Aromatic Hydrocarbon Degradation by Novel Bacteria Isolated from Burrow Sediments of Marine Benthic Macrofauna Wai Ki Chung
The University of Maine DigitalCommons@UMaine Electronic Theses and Dissertations Fogler Library 12-2001 Polycyclic aromatic hydrocarbon degradation by novel bacteria isolated from burrow sediments of marine benthic macrofauna Wai Ki Chung Follow this and additional works at: http://digitalcommons.library.umaine.edu/etd Part of the Bacteria Commons, and the Environmental Microbiology and Microbial Ecology Commons Recommended Citation Chung, Wai Ki, "Polycyclic aromatic hydrocarbon degradation by novel bacteria isolated from burrow sediments of marine benthic macrofauna" (2001). Electronic Theses and Dissertations. 521. http://digitalcommons.library.umaine.edu/etd/521 This Open-Access Dissertation is brought to you for free and open access by DigitalCommons@UMaine. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of DigitalCommons@UMaine. POLYCYCLIC AROMATIC HYDROCARBON DEGRADATION BY NOVEL BACTERIA ISOLATED FROM BURROW SEDIMENTS OF MARINE BENTHIC MACROFAUNA BY Wai Ki Chung B.Sc. The Chinese University of Hong Kong, 1990 M.Phi1. The Chinese University of Hong Kong, 1992 A THESIS Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy (in Microbiology) The Graduate School The University of Maine December, 2001 Advisory Committee: Gary King, Clare S. Darling Professor of Oceanography and Professor of Microbiology and Marine Studies, Advisor Katherine Boettcher, Assistant Professor of Microbiology Daniel Distel, Associate Professor of Biochemistry, Microbiology & Molecular Biology Jean MacRae, Assistant Professor of Civil & Environmental Engineering John Singer, Professor of Microbiology and Chair of Department of Biochemistry, Microbiology & Molecular Biology POLYCYCLIC AROMATIC HYDROCARBON DEGRADATION BY NOVEL BACTERIA ISOLATED FROM BURROW SEDIMENTS OF MARINE BENTHIC MACROFAUNA By Wai Ki Chung Thesis Advisor: Dr. -

A Novel Chlamydia Parasite of Free-Living Amoebae
CORE Metadata, citation and similar papers at core.ac.uk Provided by RERO DOC Digital Library “Candidatus Mesochlamydia elodeae” (Chlamydiae: Parachlamydiaceae), a novel chlamydia parasite of free-living amoebae Daniele Corsaro & Karl-Dieter Müller & Jost Wingender & Rolf Michel Abstract Vannella sp. isolated from waterweed Elodea sp. on the chlamydia parasite. High sequence similarity values of was found infected by a chlamydia-like organism. This organ- the 18S rDNA permitted to assign the amoeba to the species ism behaves like a parasite, causing the death through burst of Saccamoeba lacustris (Amoebozoa, Tubulinea). The bacterial its host. Once the vannellae degenerated, the parasite was endosymbiont naturally harbored by the host belonged to successfully kept in laboratory within a Saccamoeba sp. iso- Sphingomonas koreensis (Alpha-Proteobacteria). The chla- lated from the same waterweed sample, which revealed in fine mydial parasite showed a strict specificity for Saccamoeba through electron microscopy to harbor two bacterial endo- spp., being unable to infect a variety of other amoebae, in- symbionts: the chlamydial parasite we introduce and another cluding Acanthamoeba, and it was itself infected by a bacte- endosymbiont initially and naturally present in the host. riophage. Sequence similarity values of the 16S rDNA and Herein, we provide molecular-based identification of both phylogenetic analysis indicated that this strain is a new mem- the amoeba host and its two endosymbionts, with special focus ber of the family Parachlamydiaceae, for which we propose the name “Candidatus Mesochlamydia elodeae.” Introduction Chlamydiae constitute a large group of intracellular para- * D. Corsaro ( ) sites of eukaryotes, infecting amoebae and some inverte- Chlamydia Research Association (CHLAREAS), brates and vertebrates, including humans (Corsaro and 12 rue du Maconnais, 54500 Vandoeuvre-lès-Nancy, France Venditti 2004; Corsaro and Greub 2006). -
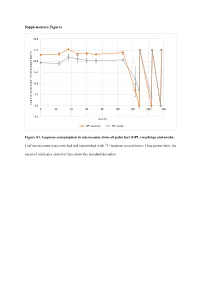
Supplementary Figures Figure S1. Isoprene Consumption In
Supplementary Figures 30.0 25.0 20.0 15.0 10.0 5.0 Isoprene concentration in the headspace (ppmv) 0.0 0 20406080100120140160 -5.0 Time (h) OPL washings OPL swabs Figure S1. Isoprene consumption in microcosms from oil palm leaf (OPL) washings and swabs. Leaf microcosms were enriched and replenished with 12C-isoprene several times. Data points show the mean of triplicates and error bars show the standard deviation. A 35.0 T1 T2 T3 30.0 25.0 20.0 15.0 10.0 5.0 Isoprene concentration in the headspace (ppm) headspace the in concentration Isoprene 0.0 -5.0 0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 Time (h) 12C-isoprene 13C-labelled isoprene B 70.0 T1 60.0 50.0 40.0 30.0 20.0 10.0 Isoprene concentration in the headspace (ppmv) 0.0 -10.0 0 20 40 60 80 100 120 140 160 180 200 220 Time (h) 12C-isoprene 13C-labelled isoprene Figure S2. Isoprene consumption in microcosms used in DNA-SIP experiments with oil palm soil (A) and leaf washings (B). Soil (A) and leaf washings (B) microcosms were enriched and replenished with 12C- or 13C-labelled isoprene. Data points show the mean of triplicates and error bars show the standard deviation. B) Leaf washings enrichment are shown up to 220 h (T1) because of variation observed in the isoprene consumption between replicates. A 12 12 C-isoprene T1 12 C-isoprene T3 L C-isoprene T2 40 35 45 L L 35 30 40 35 30 25 30 25 20 25 20 15 20 15 15 DNA concentration(ng uL-1) DNA concentrationDNA uL-1) (ng 10 DNA concentration (ng uL-1) DNA (ng concentration 10 10 5 5 5 0 0 0 1.397 1.399 1.401 1.403 -

Sphingomonas Negativus Sp. Nov., and Sphingomonas Gyeonggiense Sp
Sphingomonas Negativus sp. nov., and Sphingomonas Gyeonggiense sp. nov., Bacteria Isolated From Soil in South Korea Soohyun Maeng Seoul Women's University Yuna Park Seoul Women's University Tuvshinzaya Damdintogtokh Seoul Women's University Hyejin Oh Seoul Women's University Minji Bang Seoul Women's University Sathiyaraj Srinivasan Seoul Women's University Myung Kyum Kim ( [email protected] ) Seoul Women's University https://orcid.org/0000-0003-4098-1520 Research Article Keywords: Sphingomonadaceae, Sphingomonas, Novel species, Taxonomy Posted Date: September 2nd, 2021 DOI: https://doi.org/10.21203/rs.3.rs-644195/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/12 Abstract Two novel Gram-staining-negative bacterial strains BT553T and BT552T were isolated from soil collected in Gyeonggi province, Korea. Phylogenetic analysis using 16S rRNA gene sequences revealed that the strains BT553T and BT552T both belong to a distinct lineage in the genus Sphingomonas (family Sphingomonadaceae, order Sphingomonadales, class Alphaproteobacteria). Strain BT553T was closely related to Sphingomonas melonis DAPP-PG 224 T (98.1 % 16S rRNA gene similarity) and Sphingomonas aquatilis JSS7T (98.1%). Strain BT553T was closely related to Sphingomonas melonis DAPP-PG 224 T (98.2 %) and Sphingomonas aquatilis JSS7T (98.1%). The genome size of strain BT553T was 3,941,714 bp. Bacterial growth was observed at 10°C–30°C (optimum 25°C), pH 5.0–9.0 (optimum pH 7.0) in R2A agar and the presence of up to 2% NaCl. The genome size of strain BT552T was 4,035,561 bp.