Microbial Strain Engraftment, Persistence and Replacement After Fecal Microbiota Transplantation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Polyamine Profiles of Some Members of the Alpha Subclass of the Class Proteobacteria: Polyamine Analysis of Twenty Recently Described Genera
Microbiol. Cult. Coll. June 2003. p. 13 ─ 21 Vol. 19, No. 1 Polyamine Profiles of Some Members of the Alpha Subclass of the Class Proteobacteria: Polyamine Analysis of Twenty Recently Described Genera Koei Hamana1)*,Azusa Sakamoto1),Satomi Tachiyanagi1), Eri Terauchi1)and Mariko Takeuchi2) 1)Department of Laboratory Sciences, School of Health Sciences, Faculty of Medicine, Gunma University, 39 ─ 15 Showa-machi 3 ─ chome, Maebashi, Gunma 371 ─ 8514, Japan 2)Institute for Fermentation, Osaka, 17 ─ 85, Juso-honmachi 2 ─ chome, Yodogawa-ku, Osaka, 532 ─ 8686, Japan Cellular polyamines of 41 newly validated or reclassified alpha proteobacteria belonging to 20 genera were analyzed by HPLC. Acetic acid bacteria belonging to the new genus Asaia and the genera Gluconobacter, Gluconacetobacter, Acetobacter and Acidomonas of the alpha ─ 1 sub- group ubiquitously contained spermidine as the major polyamine. Aerobic bacteriochlorophyll a ─ containing Acidisphaera, Craurococcus and Paracraurococcus(alpha ─ 1)and Roseibium (alpha-2)contained spermidine and lacked homospermidine. New Rhizobium species, including some species transferred from the genera Agrobacterium and Allorhizobium, and new Sinorhizobium and Mesorhizobium species of the alpha ─ 2 subgroup contained homospermidine as a major polyamine. Homospermidine was the major polyamine in the genera Oligotropha, Carbophilus, Zavarzinia, Blastobacter, Starkeya and Rhodoblastus of the alpha ─ 2 subgroup. Rhodobaca bogoriensis of the alpha ─ 3 subgroup contained spermidine. Within the alpha ─ 4 sub- group, the genus Sphingomonas has been divided into four clusters, and species of the emended Sphingomonas(cluster I)contained homospermidine whereas those of the three newly described genera Sphingobium, Novosphingobium and Sphingopyxis(corresponding to clusters II, III and IV of the former Sphingomonas)ubiquitously contained spermidine. -

A Primary Assessment of the Endophytic Bacterial Community in a Xerophilous Moss (Grimmia Montana) Using Molecular Method and Cultivated Isolates
Brazilian Journal of Microbiology 45, 1, 163-173 (2014) Copyright © 2014, Sociedade Brasileira de Microbiologia ISSN 1678-4405 www.sbmicrobiologia.org.br Research Paper A primary assessment of the endophytic bacterial community in a xerophilous moss (Grimmia montana) using molecular method and cultivated isolates Xiao Lei Liu, Su Lin Liu, Min Liu, Bi He Kong, Lei Liu, Yan Hong Li College of Life Science, Capital Normal University, Haidian District, Beijing, China. Submitted: December 27, 2012; Approved: April 1, 2013. Abstract Investigating the endophytic bacterial community in special moss species is fundamental to under- standing the microbial-plant interactions and discovering the bacteria with stresses tolerance. Thus, the community structure of endophytic bacteria in the xerophilous moss Grimmia montana were esti- mated using a 16S rDNA library and traditional cultivation methods. In total, 212 sequences derived from the 16S rDNA library were used to assess the bacterial diversity. Sequence alignment showed that the endophytes were assigned to 54 genera in 4 phyla (Proteobacteria, Firmicutes, Actinobacteria and Cytophaga/Flexibacter/Bacteroids). Of them, the dominant phyla were Proteobacteria (45.9%) and Firmicutes (27.6%), the most abundant genera included Acinetobacter, Aeromonas, Enterobacter, Leclercia, Microvirga, Pseudomonas, Rhizobium, Planococcus, Paenisporosarcina and Planomicrobium. In addition, a total of 14 species belonging to 8 genera in 3 phyla (Proteo- bacteria, Firmicutes, Actinobacteria) were isolated, Curtobacterium, Massilia, Pseudomonas and Sphingomonas were the dominant genera. Although some of the genera isolated were inconsistent with those detected by molecular method, both of two methods proved that many different endophytic bacteria coexist in G. montana. According to the potential functional analyses of these bacteria, some species are known to have possible beneficial effects on hosts, but whether this is the case in G. -

Microbial Community Structure Analysis in Acer Palmatum Bark and Isolation of Novel Bacteria IAD-21 of the Phylum Abditibacteriota (Former Candidate Division FBP)
Microbial community structure analysis in Acer palmatum bark and isolation of novel bacteria IAD-21 of the phylum Abditibacteriota (former candidate division FBP) Kazuki Kobayashi1 and Hideki Aoyagi1,2 1 Division of Life Sciences and Bioengineering, Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Ibaraki, Japan 2 Faculty of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Ibaraki, Japan ABSTRACT Background: The potential of unidentified microorganisms for academic and other applications is limitless. Plants have diverse microbial communities associated with their biomes. However, few studies have focused on the microbial community structure relevant to tree bark. Methods: In this report, the microbial community structure of bark from the broad-leaved tree Acer palmatum was analyzed. Both a culture-independent approach using polymerase chain reaction (PCR) amplification and next generation sequencing, and bacterial isolation and sequence-based identification methods were used to explore the bark sample as a source of previously uncultured microorganisms. Molecular phylogenetic analyses based on PCR-amplified 16S rDNA sequences were performed. Results: At the phylum level, Proteobacteria and Bacteroidetes were relatively abundant in the A. palmatum bark. In addition, microorganisms from the phyla Acidobacteria, Gemmatimonadetes, Verrucomicrobia, Armatimonadetes, and Submitted 2 February 2019 Abditibacteriota, which contain many uncultured microbial species, existed in the Accepted 12 September 2019 A. palmatum bark. Of the 30 genera present at relatively high abundance in the bark, Published 29 October 2019 some genera belonging to the phyla mentioned were detected. A total of 70 isolates Corresponding author could be isolated and cultured using the low-nutrient agar media DR2A and Hideki Aoyagi, PE03. -

A Novel Chlamydia Parasite of Free-Living Amoebae
CORE Metadata, citation and similar papers at core.ac.uk Provided by RERO DOC Digital Library “Candidatus Mesochlamydia elodeae” (Chlamydiae: Parachlamydiaceae), a novel chlamydia parasite of free-living amoebae Daniele Corsaro & Karl-Dieter Müller & Jost Wingender & Rolf Michel Abstract Vannella sp. isolated from waterweed Elodea sp. on the chlamydia parasite. High sequence similarity values of was found infected by a chlamydia-like organism. This organ- the 18S rDNA permitted to assign the amoeba to the species ism behaves like a parasite, causing the death through burst of Saccamoeba lacustris (Amoebozoa, Tubulinea). The bacterial its host. Once the vannellae degenerated, the parasite was endosymbiont naturally harbored by the host belonged to successfully kept in laboratory within a Saccamoeba sp. iso- Sphingomonas koreensis (Alpha-Proteobacteria). The chla- lated from the same waterweed sample, which revealed in fine mydial parasite showed a strict specificity for Saccamoeba through electron microscopy to harbor two bacterial endo- spp., being unable to infect a variety of other amoebae, in- symbionts: the chlamydial parasite we introduce and another cluding Acanthamoeba, and it was itself infected by a bacte- endosymbiont initially and naturally present in the host. riophage. Sequence similarity values of the 16S rDNA and Herein, we provide molecular-based identification of both phylogenetic analysis indicated that this strain is a new mem- the amoeba host and its two endosymbionts, with special focus ber of the family Parachlamydiaceae, for which we propose the name “Candidatus Mesochlamydia elodeae.” Introduction Chlamydiae constitute a large group of intracellular para- * D. Corsaro ( ) sites of eukaryotes, infecting amoebae and some inverte- Chlamydia Research Association (CHLAREAS), brates and vertebrates, including humans (Corsaro and 12 rue du Maconnais, 54500 Vandoeuvre-lès-Nancy, France Venditti 2004; Corsaro and Greub 2006). -
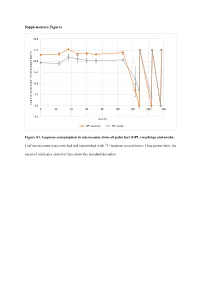
Supplementary Figures Figure S1. Isoprene Consumption In
Supplementary Figures 30.0 25.0 20.0 15.0 10.0 5.0 Isoprene concentration in the headspace (ppmv) 0.0 0 20406080100120140160 -5.0 Time (h) OPL washings OPL swabs Figure S1. Isoprene consumption in microcosms from oil palm leaf (OPL) washings and swabs. Leaf microcosms were enriched and replenished with 12C-isoprene several times. Data points show the mean of triplicates and error bars show the standard deviation. A 35.0 T1 T2 T3 30.0 25.0 20.0 15.0 10.0 5.0 Isoprene concentration in the headspace (ppm) headspace the in concentration Isoprene 0.0 -5.0 0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 Time (h) 12C-isoprene 13C-labelled isoprene B 70.0 T1 60.0 50.0 40.0 30.0 20.0 10.0 Isoprene concentration in the headspace (ppmv) 0.0 -10.0 0 20 40 60 80 100 120 140 160 180 200 220 Time (h) 12C-isoprene 13C-labelled isoprene Figure S2. Isoprene consumption in microcosms used in DNA-SIP experiments with oil palm soil (A) and leaf washings (B). Soil (A) and leaf washings (B) microcosms were enriched and replenished with 12C- or 13C-labelled isoprene. Data points show the mean of triplicates and error bars show the standard deviation. B) Leaf washings enrichment are shown up to 220 h (T1) because of variation observed in the isoprene consumption between replicates. A 12 12 C-isoprene T1 12 C-isoprene T3 L C-isoprene T2 40 35 45 L L 35 30 40 35 30 25 30 25 20 25 20 15 20 15 15 DNA concentration(ng uL-1) DNA concentrationDNA uL-1) (ng 10 DNA concentration (ng uL-1) DNA (ng concentration 10 10 5 5 5 0 0 0 1.397 1.399 1.401 1.403 -

Sphingomonas Negativus Sp. Nov., and Sphingomonas Gyeonggiense Sp
Sphingomonas Negativus sp. nov., and Sphingomonas Gyeonggiense sp. nov., Bacteria Isolated From Soil in South Korea Soohyun Maeng Seoul Women's University Yuna Park Seoul Women's University Tuvshinzaya Damdintogtokh Seoul Women's University Hyejin Oh Seoul Women's University Minji Bang Seoul Women's University Sathiyaraj Srinivasan Seoul Women's University Myung Kyum Kim ( [email protected] ) Seoul Women's University https://orcid.org/0000-0003-4098-1520 Research Article Keywords: Sphingomonadaceae, Sphingomonas, Novel species, Taxonomy Posted Date: September 2nd, 2021 DOI: https://doi.org/10.21203/rs.3.rs-644195/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/12 Abstract Two novel Gram-staining-negative bacterial strains BT553T and BT552T were isolated from soil collected in Gyeonggi province, Korea. Phylogenetic analysis using 16S rRNA gene sequences revealed that the strains BT553T and BT552T both belong to a distinct lineage in the genus Sphingomonas (family Sphingomonadaceae, order Sphingomonadales, class Alphaproteobacteria). Strain BT553T was closely related to Sphingomonas melonis DAPP-PG 224 T (98.1 % 16S rRNA gene similarity) and Sphingomonas aquatilis JSS7T (98.1%). Strain BT553T was closely related to Sphingomonas melonis DAPP-PG 224 T (98.2 %) and Sphingomonas aquatilis JSS7T (98.1%). The genome size of strain BT553T was 3,941,714 bp. Bacterial growth was observed at 10°C–30°C (optimum 25°C), pH 5.0–9.0 (optimum pH 7.0) in R2A agar and the presence of up to 2% NaCl. The genome size of strain BT552T was 4,035,561 bp. -

Bacterial Diversity and Antibiotic Resistance from the Water Source to the Tap
Bacterial diversity and antibiotic resistance from the water source to the tap Thesis submitted to the Universidade Católica Portuguesa to attain the degree of PhD in Biotechnology with specialization in Microbiology By Ivone Cristina Vaz Moreira April 2012 Bacterial diversity and antibiotic resistance from the water source to the tap Thesis submitted to the Universidade Católica Portuguesa to attain the degree of PhD in Biotechnology - with specialization in Microbiology By Ivone Cristina Vaz Moreira Under the supervision of Professor Célia Maria Manaia Rodrigues Under the co-supervision of Professor Olga Cristina Pastor Nunes April 2012 ‘‘Everything comes from water! And everything is kept alive by water!’’ J.W. von Goethe, Faust II, 1833 to all the persons who crossed my life and taught me something Abstract ABSTRACT Water is one of the most important habitats for bacteria in the environment. The continuous flux in the urban water cycle carries water through many places, dragging bacteria and numerous chemical contaminants. This makes of water one of the most important vehicles, not only for the dissemination of the chemical substances, but also for the dissemination of organisms and, consequently, the respective resistance genes in the environment. The main goal of this study was to investigate if drinking water production and distribution could represent a hotspot for the proliferation, selection or incoming of antibiotic resistant bacteria, and the likelihood of these organisms to reach the final consumer, via tap water. In order to meet this objective, the study was planned aiming the tracking of bacterial communities and individual isolates from the source to the tap. -

Sphingosinicella Humi Sp. Nov., Isolated from Arsenic- Contaminated Farmland Soil and Emended Description of the Genus Sphingosinicella
TAXONOMIC DESCRIPTION Qiao et al., Int J Syst Evol Microbiol DOI 10.1099/ijsem.0.003186 Sphingosinicella humi sp. nov., isolated from arsenic- contaminated farmland soil and emended description of the genus Sphingosinicella Zixu Qiao,1 Min Cao,1 Dan Wang,1 Shuijiao Liao1,2 and Gejiao Wang1,* Abstract A Gram-stain-negative, strictly aerobic bacterium, designated strain QZX222T, was isolated from arsenic-contaminated farmland soil. Phylogenetic analysis based on 16S rRNA gene sequences showed that strain QZX222T was clustered with Sphingosinicella vermicomposti YC7378T (97.0 %), Sphingosinicella xenopeptidilytica 3–2W4T (96.1 %), Sphingosinicella microcystinivorans Y2T (96.0 %) and Sphingosinicella soli KSL-125T (95.9 %). Compared to strain QZX222T, Spingomonas olgophenolica JCM 12082T and Sphingobium boeckii 469T had 16S rRNA gene similarities of 96.2 and 95.9 %, respectively, but they located in other phylogenetic clusters. DNA–DNA hybridization and genomic ANI values between strain QZX222T and Sphingosinicella vermicomposti DSM 21593T (KCTC 22446T) were 34.8 and 75.0 %, respectively. The genome size of strain QZX222T was 3.0 Mb including 2982 predicted genes. The strain had a DNA G+C content of 65.9 mol%. Strain QZX222T had ubiquinone Q-10 as the major respiratory quinone and homospermidine as the major polyamine. The major fatty acids T (>10 %) of strain QZX222 were C17 : 1!6c, summed feature 8 (C18 : 1!7c and/or C18 : 1!6c) and C17 : 1!8c. The polar lipids were sphingoglycolipid, phosphatidylethanolamine, phosphatidylglycerol, diphosphatidylglycerol, phosphatidylcholine and an unidentified glycolipid. Strain QZX222T could be distinguished from other Sphingosinicella strains based on the results of phylogenetic and genomic analyses, DNA–DNA hybridization, white colour colony, hydrolysis of urea, alkaline phosphatase activity, lack of phosphatidylmonomethylethanolamine, and presence of phosphatidylcholine. -

Draft Genome Sequence of the Agarase-Producing Sphingomonas Sp
DATA REPORT published: 16 March 2017 doi: 10.3389/fenvs.2017.00009 Draft Genome Sequence of the Agarase-Producing Sphingomonas sp. MCT13 Marco M. D’Andrea 1, 2*, Nagaia Ciacci 1, 3, Vincenzo Di Pilato 4, Gian M. Rossolini 1, 2, 5 and Maria C. Thaller 3 1 Department of Medical Biotechnologies, University of Siena, Siena, Italy, 2 Department of Experimental and Clinical Medicine, University of Florence, Florence, Italy, 3 Department of Biology, University of Rome Tor Vergata, Rome, Italy, 4 Department of Surgery and Translational Medicine, University of Florence, Florence, Italy, 5 Clinical Microbiology and Virology Unit, Florence Careggi University Hospital, Florence, Italy Keywords: Sphingomonas, agarase, Sphingomonadaceae, Sphingomonadales, draft genome, draft assemblies INTRODUCTION The genus Sphingomonas, originally proposed by Yabuuchi et al., was subsequently amended and is now subdivided into four genera: Sphingomonas sensu stricto, Sphingobium, Novosphingobium, and Sphingopyxis (Yabuuchi et al., 1990; Takeuchi et al., 2001). Sphingomonads have gained particular Edited by: attention for their unique abilities to degrade a variety of compounds, including pollutants Jyoti Prakash Maity, produced by industrial processes, polycyclic aromatic hydrocarbons (PAH) and dibenzofurans, and National Chung Cheng University, other toxic chemicals such as insecticides, herbicides, and heavy metals (Leys et al., 2004). Due to Taiwan these features, sphingomonads are of great interest for bioremediation purposes, and have been Reviewed by: previously -

Towards the Detection of Horizontal Gene Transfer in Metagenomic Datasets
Towards the detection of horizontal gene transfer in metagenomic datasets Weizhi Song A thesis in fulfilment of the requirements for the degree of Doctor of Philosophy School of Biotechnology and Biomolecular Sciences Faculty of Science March 2019 Surname/Family Name : Song Given Name/s : Weizhi Abbreviation for degree as give in the University calendar : PhD Faculty : Science School : Biotechnology and Biomolecular Sciences Thesis Title : Towards the detection of horizontal gene transfer in metagenomic datasets Abstract Horizontal gene transfer (HGT) is thought to be an important driving force for microbial evolution and adaptation, including the development of antibiotics resistance and niche adaptation. Metagenomics provides an opportunity to study HGT on the level of microbial communities, however, analysis method for this are currently lacking. Here, I developed three bioinformatic pipelines to aid the detection of HGT in metagenomic datasets. Firstly, Binning_refiner was developed to improve the quality of genome bins derived from metagenomic datasets through the combination of different binning programs. The results demonstrated that Binning_refiner can significantly reduce the contamination level of genome bins and increase the total size of contamination-free genome bins. Secondly, HgtSIM was developed to simulate HGT events among microbial community members with user-defined mutation levels. It was developed for testing and benchmarking pipelines for recovering HGTs from complex microbial communities. And thirdly, MetaCHIP was developed to identify HGTs at the community-level through the combination of best-match and phylogenetic approaches. Assessment of its performance on both simulated and real datasets showed that it can effectively predict HGTs with various degrees of genetic divergence from microbial communities. -

Parablastomonas Arctica Gen. Nov., Sp. Nov., Isolated from High Arctic Glacial Till
International Journal of Systematic and Evolutionary Microbiology (2015), 65, 260–266 DOI 10.1099/ijs.0.067231-0 Parablastomonas arctica gen. nov., sp. nov., isolated from high Arctic glacial till Lvzhi Ren,1 Xulu Chang,1 Fan Jiang,1 Wenjing Kan,1 Zhihao Qu,1 Xia Qiu,1 Chengxiang Fang1 and Fang Peng1,2 Correspondence 1China Center for Type Culture Collection (CCTCC), College of Life Sciences, Wuhan University, Fang Peng Wuhan 430072, PR China [email protected] 2Hubei Provincial Cooperative Innovation Center of Industrial Fermentation, Wuhan 430072, PR China A pale yellow, aerobic bacterium, strain M0-2T, was isolated from a till sample. Its taxonomic position was investigated by using a polyphasic approach. Cells were Gram-stain-negative, rod- shaped and motile. Cells reproduced by budding or asymmetrical cell division. Phylogenetic analysis based on 16S rRNA gene sequences indicated that strain M0-2T belonged to the family Sphingomonadaceae and was closely related to species of the genera Novosphingobium (96.4– 92.0 %) and Blastomonas (94.6 %), Sphingopyxis witflariensis W-50T (94.0 %), Sphingosinicella soli KSL-125T (93.6 %) and Sphingomonas astaxanthinifaciens TDMA-17T (93.5 %). Ubiquinone-10 (Q-10) was the predominant respiratory quinone. The major fatty acids were summed feature 8 (comprising C18 : 1v7c and/or C18 : 1v6c, 31.9 %), summed feature 3 (comprising C16 : 1v7c and/or C16 : 1v6c, 19.8 %) and C14 : 0 2-OH (8.9 %). Sphingoglycolipid, phosphatidylethanolamine, diphosphatidylglycerol, phosphatidylglycerol and phosphatidylcholine were the major polar lipids. Spermidine was the major polyamine observed in the cell. The genomic DNA G+C content was 47.5 mol%. -

Population Structure and Species Description of Aquatic Sphingomonadaceae
Population structure and species description of aquatic Sphingomonadaceae Dissertation at the Faculty of Biology Ludwig-Maximilians-University Munich Hong Chen 1. Reviewer: Prof. Dr. Jörg Overmann 2. Reviewer: Prof. Dr. Anton Hartmann Date of examination: 30. Jan. 2012 Publications originating from this thesis 1. Chen, H. , Jogler, M., Sikorski, J., Overmann J. Evidence for incipient speciation among sympatric subpopulations of a single phylotype of freshwater planktonic Sphingomonadaceae ISME J. , submitted 2. Chen, H. , Jogler, M., Tindall, B., Rohde, M., Busse, H.-J., Overmann J. Reclassification and amended description of Caulobacter leidyi as Sphingomonas leidyi comb. nov., and emendation of the genus Sphingomonas. Int J Syst Evol Microbiol. submitted. 3. Chen, H. , Jogler, M., Tindall, B., Rohde, M., Busse, H.-J., Overmann J. Sphingobium limneticum sp. nov., isolated from fresh lake water Starnberger See. Int J Syst Evol Microbiol. submitted. 4. Chen, H. , Jogler, M., Tindall, B., Rohde, M., Busse, H.-J., Overmann J. Sphingomonas oligotrophica sp. nov., isolated from fresh lake water Starnberger See. Int J Syst Evol Microbiol. in prep. 5. Chen, H. , Jogler, M., Tindall, B., Rohde, M., Busse, H.-J., Overmann J. Sphingobium boeckii sp. nov., isolated from fresh lake water Walchensee and reclassification of S phingomonas suberifacien as Sphingobium suberifacien . Int J Syst Evol Microbiol . submitted. Contribution of Hong Chen to the publications listed in this thesis Publication 1: Hong Chen performed the isolation and identification of all the 95 strains of Sphingomonadaceae used in the analysis; she chose and designed the primers for the 9 housekeeping genes, tested and established the PCR protocols. She also run all the molecular work such as PCR and gene sequencing, edited the sequence data; set up the clone library of gyrB gene, finished all the phenotypic characterization using the BiOLOG system.