Towards the Detection of Horizontal Gene Transfer in Metagenomic Datasets
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Polyamine Profiles of Some Members of the Alpha Subclass of the Class Proteobacteria: Polyamine Analysis of Twenty Recently Described Genera
Microbiol. Cult. Coll. June 2003. p. 13 ─ 21 Vol. 19, No. 1 Polyamine Profiles of Some Members of the Alpha Subclass of the Class Proteobacteria: Polyamine Analysis of Twenty Recently Described Genera Koei Hamana1)*,Azusa Sakamoto1),Satomi Tachiyanagi1), Eri Terauchi1)and Mariko Takeuchi2) 1)Department of Laboratory Sciences, School of Health Sciences, Faculty of Medicine, Gunma University, 39 ─ 15 Showa-machi 3 ─ chome, Maebashi, Gunma 371 ─ 8514, Japan 2)Institute for Fermentation, Osaka, 17 ─ 85, Juso-honmachi 2 ─ chome, Yodogawa-ku, Osaka, 532 ─ 8686, Japan Cellular polyamines of 41 newly validated or reclassified alpha proteobacteria belonging to 20 genera were analyzed by HPLC. Acetic acid bacteria belonging to the new genus Asaia and the genera Gluconobacter, Gluconacetobacter, Acetobacter and Acidomonas of the alpha ─ 1 sub- group ubiquitously contained spermidine as the major polyamine. Aerobic bacteriochlorophyll a ─ containing Acidisphaera, Craurococcus and Paracraurococcus(alpha ─ 1)and Roseibium (alpha-2)contained spermidine and lacked homospermidine. New Rhizobium species, including some species transferred from the genera Agrobacterium and Allorhizobium, and new Sinorhizobium and Mesorhizobium species of the alpha ─ 2 subgroup contained homospermidine as a major polyamine. Homospermidine was the major polyamine in the genera Oligotropha, Carbophilus, Zavarzinia, Blastobacter, Starkeya and Rhodoblastus of the alpha ─ 2 subgroup. Rhodobaca bogoriensis of the alpha ─ 3 subgroup contained spermidine. Within the alpha ─ 4 sub- group, the genus Sphingomonas has been divided into four clusters, and species of the emended Sphingomonas(cluster I)contained homospermidine whereas those of the three newly described genera Sphingobium, Novosphingobium and Sphingopyxis(corresponding to clusters II, III and IV of the former Sphingomonas)ubiquitously contained spermidine. -

Table S5. the Information of the Bacteria Annotated in the Soil Community at Species Level
Table S5. The information of the bacteria annotated in the soil community at species level No. Phylum Class Order Family Genus Species The number of contigs Abundance(%) 1 Firmicutes Bacilli Bacillales Bacillaceae Bacillus Bacillus cereus 1749 5.145782459 2 Bacteroidetes Cytophagia Cytophagales Hymenobacteraceae Hymenobacter Hymenobacter sedentarius 1538 4.52499338 3 Gemmatimonadetes Gemmatimonadetes Gemmatimonadales Gemmatimonadaceae Gemmatirosa Gemmatirosa kalamazoonesis 1020 3.000970902 4 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas indica 797 2.344876284 5 Firmicutes Bacilli Lactobacillales Streptococcaceae Lactococcus Lactococcus piscium 542 1.594633558 6 Actinobacteria Thermoleophilia Solirubrobacterales Conexibacteraceae Conexibacter Conexibacter woesei 471 1.385742446 7 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas taxi 430 1.265115184 8 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas wittichii 388 1.141545794 9 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas sp. FARSPH 298 0.876754244 10 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sorangium cellulosum 260 0.764953367 11 Proteobacteria Deltaproteobacteria Myxococcales Polyangiaceae Sorangium Sphingomonas sp. Cra20 260 0.764953367 12 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas panacis 252 0.741416341 -

A Primary Assessment of the Endophytic Bacterial Community in a Xerophilous Moss (Grimmia Montana) Using Molecular Method and Cultivated Isolates
Brazilian Journal of Microbiology 45, 1, 163-173 (2014) Copyright © 2014, Sociedade Brasileira de Microbiologia ISSN 1678-4405 www.sbmicrobiologia.org.br Research Paper A primary assessment of the endophytic bacterial community in a xerophilous moss (Grimmia montana) using molecular method and cultivated isolates Xiao Lei Liu, Su Lin Liu, Min Liu, Bi He Kong, Lei Liu, Yan Hong Li College of Life Science, Capital Normal University, Haidian District, Beijing, China. Submitted: December 27, 2012; Approved: April 1, 2013. Abstract Investigating the endophytic bacterial community in special moss species is fundamental to under- standing the microbial-plant interactions and discovering the bacteria with stresses tolerance. Thus, the community structure of endophytic bacteria in the xerophilous moss Grimmia montana were esti- mated using a 16S rDNA library and traditional cultivation methods. In total, 212 sequences derived from the 16S rDNA library were used to assess the bacterial diversity. Sequence alignment showed that the endophytes were assigned to 54 genera in 4 phyla (Proteobacteria, Firmicutes, Actinobacteria and Cytophaga/Flexibacter/Bacteroids). Of them, the dominant phyla were Proteobacteria (45.9%) and Firmicutes (27.6%), the most abundant genera included Acinetobacter, Aeromonas, Enterobacter, Leclercia, Microvirga, Pseudomonas, Rhizobium, Planococcus, Paenisporosarcina and Planomicrobium. In addition, a total of 14 species belonging to 8 genera in 3 phyla (Proteo- bacteria, Firmicutes, Actinobacteria) were isolated, Curtobacterium, Massilia, Pseudomonas and Sphingomonas were the dominant genera. Although some of the genera isolated were inconsistent with those detected by molecular method, both of two methods proved that many different endophytic bacteria coexist in G. montana. According to the potential functional analyses of these bacteria, some species are known to have possible beneficial effects on hosts, but whether this is the case in G. -

Sphingobium Estronivorans Sp. Nov. and Sphingobium Bisphenolivorans Sp
TAXONOMIC DESCRIPTION Qin et al., Int. J. Syst. Evol. Microbiol. 2020;70:1822–1829 DOI 10.1099/ijsem.0.003978 Sphingobium estronivorans sp. nov. and Sphingobium bisphenolivorans sp. nov., isolated from a wastewater treatment plant Dan Qin1,2, Cong Ma3, Min Lv4 and Chang- Ping Yu1,5,* ABSTRACT Two Gram- stain- negative, aerobic, motile and rod- shaped bacteria, one designated as strain AXBT, capable of degrading estro- gens, and another, YL23T, capable of degrading estrogen and bisphenol A, were isolated from activated sludge in Xiamen City, PR China. The optimum temperature and pH of both strains were 25–35 °C and pH 7.0–8.0. While strain AXBT could tolerate 3 % (w/v) NaCl, YL23T could only grow between 0–1 % (w/v) NaCl. They contained ubiquinone-10 as the major quinone, spermidine as the major polyamine, summed feature 8 (comprising C18:1ω6c and/or C18:1ω7c) as the major fatty acids and diphosphatidyl- glycerol, phosphatidylcholine, phosphatidyldimethylethanolamine, phosphatidylethanolamine, phosphatidylglycerol and sphin- goglycolipid as the major polar lipids. The DNA G+C contents of strains AXBT and YL23T were 63.6 and 63.7 mol%, respectively. Based on the results of 16S rRNA gene sequence analysis, strains AXBT and YL23T belonged to the genus Sphingobium. Strain AXBT was most closely related to Sphingobium chlorophenolicum NBRC 16172T (97.5 %) and Sphingobium chungbukense DJ77T (97.2 %), and strain YL23T was most closely related to S. chlorophenolicum NBRC 16172T (97.4 %) and S. quisquiliarum P25T (97.1 %). Average nucleotide identity values between these two strains and S. chlorophenolicum NBRC 16172T, S. chungbuke- nse DJ77T, Sphingobium chinhatense IP26T, Sphingobium quisquiliarum P25T and Sphingobium japonicum UT26ST were from 80.7 to 85.8 %. -

Downloaded from the NCBI Database on December 2017
G C A T T A C G G C A T genes Article Genomic Analysis of γ-Hexachlorocyclohexane-Degrading Sphingopyxis lindanitolerans WS5A3p Strain in the Context of the Pangenome of Sphingopyxis Michal A. Kaminski 1 , Adam Sobczak 1,2, Andrzej Dziembowski 1,2 and Leszek Lipinski 1,2,* 1 Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawinskiego 5a, 02-106 Warsaw, Poland 2 Institute of Genetics and Biotechnology, Faculty of Biology, University of Warsaw, Pawinskiego 5a, 02-106 Warsaw, Poland * Correspondence: [email protected] Received: 29 July 2019; Accepted: 2 September 2019; Published: 6 September 2019 Abstract: Sphingopyxis inhabit diverse environmental niches, including marine, freshwater, oceans, soil and anthropogenic sites. The genus includes 20 phylogenetically distinct, valid species, but only a few with a sequenced genome. In this work, we analyzed the nearly complete genome of the newly described species, Sphingopyxis lindanitolerans, and compared it to the other available Sphingopyxis genomes. The genome included 4.3 Mbp in total and consists of a circular chromosome, and two putative plasmids. Among the identified set of lin genes responsible for γ-hexachlorocyclohexane pesticide degradation, we discovered a gene coding for a new isoform of the LinA protein. The significant potential of this species in the remediation of contaminated soil is also correlated with the fact that its genome encodes a higher number of enzymes potentially involved in aromatic compound degradation than for most other Sphingopyxis strains. Additional analysis of 44 Sphingopyxis representatives provides insights into the pangenome of Sphingopyxis and revealed a core of 734 protein clusters and between four and 1667 unique proteins per genome. -

Sphingobium Cupriresistens Sp. Nov., a Copper- Resistant Bacterium Isolated from Copper Mine Soil, and Emended Description of the Genus Sphingobium
%paper no. ije040865 charlesworth ref: ije040865& New Taxa - Proteobacteria International Journal of Systematic and Evolutionary Microbiology (2013), 63, 000–000 DOI 10.1099/ijs.0.040865-0 Sphingobium cupriresistens sp. nov., a copper- resistant bacterium isolated from copper mine soil, and emended description of the genus Sphingobium Liqiong Li,3 Hongliang Liu,3 Zunji Shi and Gejiao Wang Correspondence State Key Laboratory of Agricultural Microbiology, College of Life Science and Technology, Gejiao Wang Huazhong Agricultural University, Wuhan, 430070, PR China [email protected] or [email protected] A Gram-negative, aerobic, copper-resistant bacterium, designated strain CU4T, was isolated from copper mine soil in Daye, China. Phylogenetic analysis based on 16S rRNA gene sequences showed highest similarity to Sphingobium rhizovicinum CC-FH12-1T (98.4 %), followed by Sphingobium francense Sp+T (97.2 %), Sphingobium japonicum UT26T (97.1 %), Sphingobium abikonense NBRC 16140T (97.0 %), Sphingobium xenophagum DSM 6383T (96.9 %) and Sphingobium yanoikuyae DSM 7462T (95.5 %). The major fatty acids (.5 %) were summed feature 7 (C18 : 1v7c,C18 : 1v9t and/or C18 : 1v12t), summed feature 4 (C16 : 1v7c and/or iso- C15 : 0 2-OH), C16 : 0 and C14 : 0 2-OH, and the predominant quinone was ubiquinone Q-10. Spermidine was the major polyamine component. The major polar lipids were diphosphatidyl- glycerol, phosphatidylethanolamine, phosphatidylglycerol, sphingoglycolipid, phosphatidyldi- methylethanolamine and phosphatidylcholine. The genomic DNA G+C content of strain CU4T was 64.9 mol%. Comparison of DNA–DNA hybridization, phenotypic and chemotaxonomic characteristics between strain CU4T and phylogenetically related strains revealed that the new isolate represents a novel species of the genus Sphingobium, for which the name Sphingobium cupriresistens sp. -

Genome Organization of Sphingobium Indicum B90A: an Archetypal Hexachlorocyclohexane (HCH) Degrading Genotype
Genome Organization of Sphingobium indicum B90A: An Archetypal Hexachlorocyclohexane (HCH) Degrading Genotype Helianthous Vermaa, Abhay Bajaja, Roshan Kumara, Jasvinder Kaura, Shailly Ananda, Namita Nayyara, Akshita Puria,c, Yogendra Singhb, J. P. Khuranac and Rup Lala* *Author for correspondence: Prof. Rup Lal, e-mail: [email protected], aRoom No. 115, Molecular Biology Laboratory, Department of Zoology, University of Delhi, Delhi 110007, India. bRoom No. 112, Bacterial Pathogenesis Laboratory, Department of Zoology, University of Delhi, Delhi 110007, India. cInterdisciplinary Centre for Plant Genomics & Department of Plant Molecular Biology, University of Delhi South Campus, New Delhi, India. Data Deposition: Sequence data deposited in NCBI database under GenBank Accession numbers: CP013070.1, CP013071.1, CP013072.1 and CP013073.1. © The Author(s) 2017. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution. This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/), which permits non-commercial re-use, distribution, and reproduction in any medium, provided the original work is properly cited. For commercial re-use, please contact [email protected] Abstract. Among sphingomonads, Sphingobium indicum B90A is a widely investigated for its ability to degrade a manmade pesticide, γ-hexachlorocyclohexane (γ-HCH) and its isomers (α-, β-, δ- and ε-HCH). In this study, complete genome of strain B90A was constructed using Single Molecule Real Time Sequencing (SMRT) and Illumina platform. The complete genome revealed that strain B90A harbors four replicons: one chromosome (3,654,322 bp) and three plasmids designated as pSRL1 (139,218 bp), pSRL2 (108,430 bp) and pSRL3 (43,761 bp). -

Microbial Community Structure Analysis in Acer Palmatum Bark and Isolation of Novel Bacteria IAD-21 of the Phylum Abditibacteriota (Former Candidate Division FBP)
Microbial community structure analysis in Acer palmatum bark and isolation of novel bacteria IAD-21 of the phylum Abditibacteriota (former candidate division FBP) Kazuki Kobayashi1 and Hideki Aoyagi1,2 1 Division of Life Sciences and Bioengineering, Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Ibaraki, Japan 2 Faculty of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Ibaraki, Japan ABSTRACT Background: The potential of unidentified microorganisms for academic and other applications is limitless. Plants have diverse microbial communities associated with their biomes. However, few studies have focused on the microbial community structure relevant to tree bark. Methods: In this report, the microbial community structure of bark from the broad-leaved tree Acer palmatum was analyzed. Both a culture-independent approach using polymerase chain reaction (PCR) amplification and next generation sequencing, and bacterial isolation and sequence-based identification methods were used to explore the bark sample as a source of previously uncultured microorganisms. Molecular phylogenetic analyses based on PCR-amplified 16S rDNA sequences were performed. Results: At the phylum level, Proteobacteria and Bacteroidetes were relatively abundant in the A. palmatum bark. In addition, microorganisms from the phyla Acidobacteria, Gemmatimonadetes, Verrucomicrobia, Armatimonadetes, and Submitted 2 February 2019 Abditibacteriota, which contain many uncultured microbial species, existed in the Accepted 12 September 2019 A. palmatum bark. Of the 30 genera present at relatively high abundance in the bark, Published 29 October 2019 some genera belonging to the phyla mentioned were detected. A total of 70 isolates Corresponding author could be isolated and cultured using the low-nutrient agar media DR2A and Hideki Aoyagi, PE03. -

Sphingobium Bisphenolivorans Sp
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Institutional Repository of Yantai Institute of Coastal Zone Research, CAS TAXONOMIC DESCRIPTION Qin et al., Int. J. Syst. Evol. Microbiol. 2020;70:1822–1829 DOI 10.1099/ijsem.0.003978 Sphingobium estronivorans sp. nov. and Sphingobium bisphenolivorans sp. nov., isolated from a wastewater treatment plant Dan Qin1,2, Cong Ma3, Min Lv4 and Chang- Ping Yu1,5,* ABSTRACT Two Gram- stain- negative, aerobic, motile and rod- shaped bacteria, one designated as strain AXBT, capable of degrading estro- gens, and another, YL23T, capable of degrading estrogen and bisphenol A, were isolated from activated sludge in Xiamen City, PR China. The optimum temperature and pH of both strains were 25–35 °C and pH 7.0–8.0. While strain AXBT could tolerate 3 % (w/v) NaCl, YL23T could only grow between 0–1 % (w/v) NaCl. They contained ubiquinone-10 as the major quinone, spermidine as the major polyamine, summed feature 8 (comprising C18:1ω6c and/or C18:1ω7c) as the major fatty acids and diphosphatidyl- glycerol, phosphatidylcholine, phosphatidyldimethylethanolamine, phosphatidylethanolamine, phosphatidylglycerol and sphin- goglycolipid as the major polar lipids. The DNA G+C contents of strains AXBT and YL23T were 63.6 and 63.7 mol%, respectively. Based on the results of 16S rRNA gene sequence analysis, strains AXBT and YL23T belonged to the genus Sphingobium. Strain AXBT was most closely related to Sphingobium chlorophenolicum NBRC 16172T (97.5 %) and Sphingobium chungbukense DJ77T (97.2 %), and strain YL23T was most closely related to S. -

Sphingobium Czechense Sp. Nov., Isolated from a Hexachlorocyclohexane Dump Site
International Journal of Systematic and Evolutionary Microbiology (2013), 63, 723–728 DOI 10.1099/ijs.0.039396-0 Sphingobium czechense sp. nov., isolated from a hexachlorocyclohexane dump site Neha Niharika,13 Hana Moskalikova,23 Jasvinder Kaur,13 Fazlurrahman Khan,3 Miroslava Sedlackova,4 Ales Hampl,4 Jiri Damborsky,2,5 Zbynek Prokop5 and Rup Lal1 Correspondence 1Molecular Biology Laboratory, Department of Zoology, University of Delhi, Delhi – 110007, India Zbynek Prokop 2International Clinical Research Center, St. Anne’s University Hospital Brno, Pekarska 53, [email protected] 656 91 Brno, Czech Republic Rup Lal 3 [email protected] IMTECH – Institute of Microbial Technology, Sector-39A, Chandigarh – 160036, India 4Department of Histology and Embryology, Faculty of Medicine, Masaryk University, 625 00 Brno, Czech Republic 5Loschmidt Laboratories and Research Centre for Toxic Compounds in the Environment, Faculty of Science, Masaryk University, 628 00 Brno, Czech Republic A yellow-pigmented bacterial strain, designated LL01T, was isolated from hexachlorocyclohexane (HCH)-contaminated soil at Spolana Neratovice, a former Czech producer of lindane. A neighbour-joining tree based on 16S rRNA gene sequences showed that strain LL01T occupied a distinct phylogenetic position in the Sphingobium cluster, showing highest similarity to Sphingobium rhizovicinum CC-FH12-1T (98.5 %). The DNA G+C content of strain LL01T was 66.1 mol%. The predominant respiratory pigment was ubiquinone Q-10. The polar lipid profile of strain LL01T also corresponded to those reported for other Sphingobium species (phosphatidyl- ethanolamine, diphosphatidylglycerol, phosphatidylcholine, phosphatidylglycerol, phosphatidyl- monomethylethanolamine, phosphatidyldimethylethanolamine, sphingoglycolipids), supporting its identification as a member of the genus Sphingobium. Spermidine was the major polyamine observed. -

A Novel Chlamydia Parasite of Free-Living Amoebae
CORE Metadata, citation and similar papers at core.ac.uk Provided by RERO DOC Digital Library “Candidatus Mesochlamydia elodeae” (Chlamydiae: Parachlamydiaceae), a novel chlamydia parasite of free-living amoebae Daniele Corsaro & Karl-Dieter Müller & Jost Wingender & Rolf Michel Abstract Vannella sp. isolated from waterweed Elodea sp. on the chlamydia parasite. High sequence similarity values of was found infected by a chlamydia-like organism. This organ- the 18S rDNA permitted to assign the amoeba to the species ism behaves like a parasite, causing the death through burst of Saccamoeba lacustris (Amoebozoa, Tubulinea). The bacterial its host. Once the vannellae degenerated, the parasite was endosymbiont naturally harbored by the host belonged to successfully kept in laboratory within a Saccamoeba sp. iso- Sphingomonas koreensis (Alpha-Proteobacteria). The chla- lated from the same waterweed sample, which revealed in fine mydial parasite showed a strict specificity for Saccamoeba through electron microscopy to harbor two bacterial endo- spp., being unable to infect a variety of other amoebae, in- symbionts: the chlamydial parasite we introduce and another cluding Acanthamoeba, and it was itself infected by a bacte- endosymbiont initially and naturally present in the host. riophage. Sequence similarity values of the 16S rDNA and Herein, we provide molecular-based identification of both phylogenetic analysis indicated that this strain is a new mem- the amoeba host and its two endosymbionts, with special focus ber of the family Parachlamydiaceae, for which we propose the name “Candidatus Mesochlamydia elodeae.” Introduction Chlamydiae constitute a large group of intracellular para- * D. Corsaro ( ) sites of eukaryotes, infecting amoebae and some inverte- Chlamydia Research Association (CHLAREAS), brates and vertebrates, including humans (Corsaro and 12 rue du Maconnais, 54500 Vandoeuvre-lès-Nancy, France Venditti 2004; Corsaro and Greub 2006). -
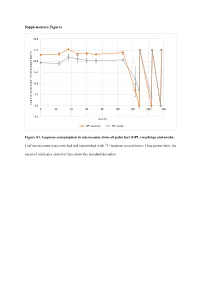
Supplementary Figures Figure S1. Isoprene Consumption In
Supplementary Figures 30.0 25.0 20.0 15.0 10.0 5.0 Isoprene concentration in the headspace (ppmv) 0.0 0 20406080100120140160 -5.0 Time (h) OPL washings OPL swabs Figure S1. Isoprene consumption in microcosms from oil palm leaf (OPL) washings and swabs. Leaf microcosms were enriched and replenished with 12C-isoprene several times. Data points show the mean of triplicates and error bars show the standard deviation. A 35.0 T1 T2 T3 30.0 25.0 20.0 15.0 10.0 5.0 Isoprene concentration in the headspace (ppm) headspace the in concentration Isoprene 0.0 -5.0 0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 Time (h) 12C-isoprene 13C-labelled isoprene B 70.0 T1 60.0 50.0 40.0 30.0 20.0 10.0 Isoprene concentration in the headspace (ppmv) 0.0 -10.0 0 20 40 60 80 100 120 140 160 180 200 220 Time (h) 12C-isoprene 13C-labelled isoprene Figure S2. Isoprene consumption in microcosms used in DNA-SIP experiments with oil palm soil (A) and leaf washings (B). Soil (A) and leaf washings (B) microcosms were enriched and replenished with 12C- or 13C-labelled isoprene. Data points show the mean of triplicates and error bars show the standard deviation. B) Leaf washings enrichment are shown up to 220 h (T1) because of variation observed in the isoprene consumption between replicates. A 12 12 C-isoprene T1 12 C-isoprene T3 L C-isoprene T2 40 35 45 L L 35 30 40 35 30 25 30 25 20 25 20 15 20 15 15 DNA concentration(ng uL-1) DNA concentrationDNA uL-1) (ng 10 DNA concentration (ng uL-1) DNA (ng concentration 10 10 5 5 5 0 0 0 1.397 1.399 1.401 1.403