Life Histories of Bacteria: Genomic Foundations and Ecological Implications
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Novel Bacterial Lineages Associated with Boreal Moss Species Hannah
bioRxiv preprint doi: https://doi.org/10.1101/219659; this version posted November 16, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Novel bacterial lineages associated with boreal moss species 2 Hannah Holland-Moritz1,2*, Julia Stuart3, Lily R. Lewis4, Samantha Miller3, Michelle C. Mack3, Stuart 3 F. McDaniel4, Noah Fierer1,2* 4 Affiliations: 5 1Cooperative Institute for Research in Environmental Sciences, University of Colorado at Boulder, 6 Boulder, CO, USA 7 2Department of Ecology and Evolutionary Biology, University of Colorado at Boulder, Boulder, CO, 8 USA 9 3Center for Ecosystem Science and Society, Northern Arizona University, Flagstaff, AZ USA 10 4Department of Biology, University of Florida, Gainesville, FL 32611-8525, USA 11 *Corresponding Author 12 13 Abstract 14 Mosses are critical components of boreal ecosystems where they typically account for a large 15 proportion of net primary productivity and harbor diverse bacterial communities that can be the major 16 source of biologically-fixed nitrogen in these ecosystems. Despite their ecological importance, we have 17 limited understanding of how microbial communities vary across boreal moss species and the extent to 18 which local environmental conditions may influence the composition of these bacterial communities. 19 We used marker gene sequencing to analyze bacterial communities associated with eight boreal moss 20 species collected near Fairbanks, AK USA. We found that host identity was more important than site in 21 determining bacterial community composition and that mosses harbor diverse lineages of potential N2- 22 fixers as well as an abundance of novel taxa assigned to understudied bacterial phyla (including 23 candidate phylum WPS-2). -

Genomic Analysis of Family UBA6911 (Group 18 Acidobacteria)
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.09.439258; this version posted April 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 2 Genomic analysis of family UBA6911 (Group 18 3 Acidobacteria) expands the metabolic capacities of the 4 phylum and highlights adaptations to terrestrial habitats. 5 6 Archana Yadav1, Jenna C. Borrelli1, Mostafa S. Elshahed1, and Noha H. Youssef1* 7 8 1Department of Microbiology and Molecular Genetics, Oklahoma State University, Stillwater, 9 OK 10 *Correspondence: Noha H. Youssef: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2021.04.09.439258; this version posted April 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 11 Abstract 12 Approaches for recovering and analyzing genomes belonging to novel, hitherto unexplored 13 bacterial lineages have provided invaluable insights into the metabolic capabilities and 14 ecological roles of yet-uncultured taxa. The phylum Acidobacteria is one of the most prevalent 15 and ecologically successful lineages on earth yet, currently, multiple lineages within this phylum 16 remain unexplored. Here, we utilize genomes recovered from Zodletone spring, an anaerobic 17 sulfide and sulfur-rich spring in southwestern Oklahoma, as well as from multiple disparate soil 18 and non-soil habitats, to examine the metabolic capabilities and ecological role of members of 19 the family UBA6911 (group18) Acidobacteria. -

Biorremediación De Metales Pesados Por Sulfidogénesis Utilizando Comunidades Y Microorganismos Sulfato-Reductores
FACULTAD DE CIENCIAS EXACTAS DEPARTAMENTO DE QUÍMICA BIORREMEDIACIÓN DE METALES PESADOS POR SULFIDOGÉNESIS UTILIZANDO COMUNIDADES Y MICROORGANISMOS SULFATO-REDUCTORES Trabajo de Tesis Doctoral Graciana Willis Poratti Director: Dr. Edgardo Donati Año 2016 Este Trabajo de Tesis Doctoral es presentado para optar al grado de Doctor de la Facultad de Ciencias Exactas Fue realizado en CINDEFI (CCT La Plata-CONICET, UNLP) A Mamá, Papá y Juan, por acompañarme siempre Reconocimientos A la Facultad de Ciencias Exactas de la Universidad Nacional de La Plata y al Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) por haber hecho posible la realización del presente Trabajo de Tesis Doctoral. Al CINDEFI (Centro de Investigación en Fermentaciones Industriales) y a toda la gente que lo conforma por haberme brindado el lugar físico para la realización del presente trabajo. Al Prof. Dr. David Barrie Johnson (Bangor Acidophile Research Team) de la Universidad de Bangor, Gales por permitirme realizar parte del trabajo de Tesis Doctoral en su laboratorio. A los Dres. Goh Kian Mau de la Universiti Teknologi, Malaysia y Kok-Gan Chan de la University of Malaysa, por la colaboración en la secuenciación del genoma de la cepa LMa1. Al Ente Provincial de Termas de Neuquén (EPROTEN) por permitir la toma de muestra en el Parque Geotermal Copahue. Al grupo de la Dra. Alejandra Giaveno perteneciente al IDEPA (CONICET, UNCo) y a la Facultad de Ingeniería de la Universidad Nacional del Comahue, por la organización de las campañas de muestreo a la zona de Copahue. Agradecimientos A mi director Dr. Edgardo Donati, por haber depositado su confianza en la realización de este trabajo, por dirigirme y haberme dado la oportunidad de viajar y formarme. -

Tree-Aggregated Predictive Modeling of Microbiome Data
www.nature.com/scientificreports OPEN Tree‑aggregated predictive modeling of microbiome data Jacob Bien1, Xiaohan Yan2, Léo Simpson3,4 & Christian L. Müller4,5,6* Modern high‑throughput sequencing technologies provide low‑cost microbiome survey data across all habitats of life at unprecedented scale. At the most granular level, the primary data consist of sparse counts of amplicon sequence variants or operational taxonomic units that are associated with taxonomic and phylogenetic group information. In this contribution, we leverage the hierarchical structure of amplicon data and propose a data‑driven and scalable tree‑guided aggregation framework to associate microbial subcompositions with response variables of interest. The excess number of zero or low count measurements at the read level forces traditional microbiome data analysis workfows to remove rare sequencing variants or group them by a fxed taxonomic rank, such as genus or phylum, or by phylogenetic similarity. By contrast, our framework, which we call trac (tree‑aggregation of compositional data), learns data‑adaptive taxon aggregation levels for predictive modeling, greatly reducing the need for user‑defned aggregation in preprocessing while simultaneously integrating seamlessly into the compositional data analysis framework. We illustrate the versatility of our framework in the context of large‑scale regression problems in human gut, soil, and marine microbial ecosystems. We posit that the inferred aggregation levels provide highly interpretable taxon groupings that can help microbiome researchers gain insights into the structure and functioning of the underlying ecosystem of interest. Microbial communities populate all major environments on earth and signifcantly contribute to the total plan- etary biomass. Current estimates suggest that a typical human-associated microbiome consists of ∼ 1013 bacteria1 and that marine bacteria and protists contribute to as much as 70% of the total marine biomass2. -

Bibliography
Bibliography Abella, C.A., X.P. Cristina, A. Martinez, I. Pibernat and X. Vila. 1998. on moderate concentrations of acetate: production of single cells. Two new motile phototrophic consortia: "Chlorochromatium lunatum" Appl. Microbiol. Biotechnol. 35: 686-689. and "Pelochromatium selenoides". Arch. Microbiol. 169: 452-459. Ahring, B.K, P. Westermann and RA. Mah. 1991b. Hydrogen inhibition Abella, C.A and LJ. Garcia-Gil. 1992. Microbial ecology of planktonic of acetate metabolism and kinetics of hydrogen consumption by Me filamentous phototrophic bacteria in holomictic freshwater lakes. Hy thanosarcina thermophila TM-I. Arch. Microbiol. 157: 38-42. drobiologia 243-244: 79-86. Ainsworth, G.C. and P.H.A Sheath. 1962. Microbial Classification: Ap Acca, M., M. Bocchetta, E. Ceccarelli, R Creti, KO. Stetter and P. Cam pendix I. Symp. Soc. Gen. Microbiol. 12: 456-463. marano. 1994. Updating mass and composition of archaeal and bac Alam, M. and D. Oesterhelt. 1984. Morphology, function and isolation terial ribosomes. Archaeal-like features of ribosomes from the deep of halobacterial flagella. ]. Mol. Biol. 176: 459-476. branching bacterium Aquifex pyrophilus. Syst. Appl. Microbiol. 16: 629- Albertano, P. and L. Kovacik. 1994. Is the genus LeptolynglYya (Cyano 637. phyte) a homogeneous taxon? Arch. Hydrobiol. Suppl. 105: 37-51. Achenbach-Richter, L., R Gupta, KO. Stetter and C.R Woese. 1987. Were Aldrich, H.C., D.B. Beimborn and P. Schönheit. 1987. Creation of arti the original eubacteria thermophiles? Syst. Appl. Microbiol. 9: 34- factual internal membranes during fixation of Methanobacterium ther 39. moautotrophicum. Can.]. Microbiol. 33: 844-849. Adams, D.G., D. Ashworth and B. -

Microbial Communities of Polymetallic Deposits' Acidic
Microbial Communities of Polymetallic Deposits’ Acidic Ecosystems of ANGOR UNIVERSITY Continental Climatic Zone With High Temperature Contrasts Gavrilov, Sergei N.; Korzhenkov, Aleksei A.; Kublanov, Ilya V.; Bargiela, Rafael; Zamana, Leonid V.; Toshchakov, Stepan V.; Golyshin, Peter; Golyshina, Olga Frontiers in Microbiology PRIFYSGOL BANGOR / B Published: 17/07/2019 Publisher's PDF, also known as Version of record Cyswllt i'r cyhoeddiad / Link to publication Dyfyniad o'r fersiwn a gyhoeddwyd / Citation for published version (APA): Gavrilov, S. N., Korzhenkov, A. A., Kublanov, I. V., Bargiela, R., Zamana, L. V., Toshchakov, S. V., Golyshin, P., & Golyshina, O. (2019). Microbial Communities of Polymetallic Deposits’ Acidic Ecosystems of Continental Climatic Zone With High Temperature Contrasts. Frontiers in Microbiology. Hawliau Cyffredinol / General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. 01. Oct. 2021 ORIGINAL RESEARCH published: 17 July 2019 doi: 10.3389/fmicb.2019.01573 Microbial Communities of Polymetallic Deposits’ Acidic Ecosystems of Continental Climatic Zone With High Temperature Contrasts Sergey N. -
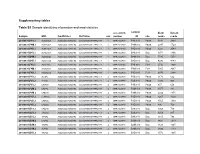
Supplementary Tables Table S1 Sample Identifying Information And
Supplementary tables Table S1 Sample identifying information and read statistics accession sample #raw #clean Sample MID FwdPrimer RvPrimer run number ID site reads reads 20100831.P4S.1 ACACGAGA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 P4S.910 Palsa 3831 2893 20100831.P4M.1 ACATAGCA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 P4M.910 Palsa 2047 1729 20100831.P4D.1 ACGATACA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 P4D.910 Palsa 3221 2543 20100831.B4S.1 ACTGCTCA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 B4S.910 Bog 1971 1366 20100831.B4M.1 AGCAGCGC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 B4M.910 Bog 5530 5251 20100831.B4D.1 AGCGTGCA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 B4D.910 Bog 4296 4143 20100831.F4S.1 AGTCTAGC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 F4S.910 Fen* 2716 1890 20100831.F4M.1 ATGACTGA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 F4M.910 Fen* 5005 3987 20100831.F4D.1 ATGCACGC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 F4D.910 Fen* 2870 2384 20100901.P1S.2 ACTGAT ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P1S.910 Palsa 1170 662 20100901.P2S.2 ATGTGT ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P2S.910 Palsa 1320 923 20100901.P3S.2 CACAGT ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P3S.910 Palsa 857 656 20100901.P2M.2 CACTAC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P2M.910 Palsa 1077 881 20100901.P3M.2 CAGCAT ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P3M.910 Palsa 2032 1474 20100901.P1D.2 CTAGAGC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT -

Pyrosequencing Investigation Into the Bacterial Community in Permafrost Soils Along the China-Russia Crude Oil Pipeline (CRCOP)
Pyrosequencing Investigation into the Bacterial Community in Permafrost Soils along the China-Russia Crude Oil Pipeline (CRCOP) Sizhong Yang1*, Xi Wen2, Huijun Jin1, Qingbai Wu1 1 State Key Laboratory of Frozen Soil Engineering (SKLFSE), Cold and Arid Regions Environmental and Engineering Research Institute (CAREERI), Chinese Academy of Sciences, Lanzhou, China, 2 College of Electrical Engineering, Northwest University for Nationalities, Lanzhou, China Abstract The China-Russia Crude Oil Pipeline (CRCOP) goes through 441 km permafrost soils in northeastern China. The bioremediation in case of oil spills is a major concern. So far, little is known about the indigenous bacteria inhabiting in the permafrost soils along the pipeline. A pilot 454 pyrosequencing analysis on the communities from four selected sites which possess high environment risk along the CRCOP is herein presented. The results reveal an immense bacterial diversity than previously anticipated. A total of 14448 OTUs with 84834 reads are identified, which could be assigned into 39 different phyla, and 223 families or 386 genera. Only five phyla sustain a mean OTU abundance more than 5% in all the samples, but they altogether account for 85.08% of total reads. Proteobacteria accounts for 41.65% of the total OTUs or 45% of the reads across all samples, and its proportion generally increases with soil depth, but OTUs numerically decline. Among Proteobacteria, the abundance of Beta-, Alpha-, Delta- and Gamma- subdivisions average to 38.7% (2331 OTUs), 37.5% (2257 OTUs), 10.35% (616 OTUs), and 6.21% (374 OTUs), respectively. Acidobacteria (esp. Acidobacteriaceae), Actinobacteria (esp. Intrasporangiaceae), Bacteroidetes (esp. Sphingobacteria and Flavobacteria) and Chloroflexi (esp. -

Und Buttersäure-Bildenden Bakterien Aus Biogasanlagen
Johannes Gutenberg-Universität Mainz Institut für Mikrobiologie und Weinforschung Isolierung von Essigsäure-, Propionsäure- und Buttersäure-bildenden Bakterien aus Biogasanlagen Dissertation Zur Erlangung des Grades Doktor der Naturwissenschaften Am Fachbereich Biologie Der Johannes Gutenberg-Universität Mainz Katharina Gabriela Cibis geboren am 19. Juni 1987 in Worms Mainz, 2015 Dekan: 1. Berichterstatter: 2. Berichterstatter: Tag der mündlichen Prüfung: 3. Dezember 2015 Die Ergebnisse dieser Arbeit sind teilweise in folgenden Publikationen und Posterpräsentatio- nen veröffentlicht oder sind zur Publikation eingereicht: INHALTSVERZEICHNIS Inhaltsverzeichnis 1. Einleitung 1 1.1 Biogasanlagen: Gewinnung von Energie und Forschungsschwerpunkt 1 1.2 Mikrobiologische Prozesse in Biogasanlagen 4 1.3 Aufbau und Betrieb von Biogasanlagen 8 1.4 Substrate für den Betrieb von Biogasanlagen 12 1.5 Mikrobielle Bildung von Essigsäure, Propionsäure und Buttersäure 13 1.6 Ziele der Arbeit 23 2. Material und Methoden 25 2.1 Chemikalien und Gase 25 2.2 Biochemikalien, Enzyme und Kits 27 2.3 Geräte und Hilfsmittel 28 2.4 Verbrauchsmaterialien 30 2.5 Mikrobiologische Methoden 31 2.5.1 Organismen 31 2.5.2 Untersuchte Biogasanlagen 31 2.5.3 Nährmedien zur Isolierung und Kultivierung von Säure-bildenden Bakterien 33 2.5.4 Methoden zur Isolierung von Säure-bildenden Bakterien 37 2.5.5 Physiologische Charakterisierung der Isolate 39 2.6 Analytische Methode: Hochleistungsflüssigkeitschromatographie 41 2.7 Molekularbiologische Methoden 42 2.7.1 DNA-Isolierung aus -

Contents Topic 1. Introduction to Microbiology. the Subject and Tasks
Contents Topic 1. Introduction to microbiology. The subject and tasks of microbiology. A short historical essay………………………………………………………………5 Topic 2. Systematics and nomenclature of microorganisms……………………. 10 Topic 3. General characteristics of prokaryotic cells. Gram’s method ………...45 Topic 4. Principles of health protection and safety rules in the microbiological laboratory. Design, equipment, and working regimen of a microbiological laboratory………………………………………………………………………….162 Topic 5. Physiology of bacteria, fungi, viruses, mycoplasmas, rickettsia……...185 TOPIC 1. INTRODUCTION TO MICROBIOLOGY. THE SUBJECT AND TASKS OF MICROBIOLOGY. A SHORT HISTORICAL ESSAY. Contents 1. Subject, tasks and achievements of modern microbiology. 2. The role of microorganisms in human life. 3. Differentiation of microbiology in the industry. 4. Communication of microbiology with other sciences. 5. Periods in the development of microbiology. 6. The contribution of domestic scientists in the development of microbiology. 7. The value of microbiology in the system of training veterinarians. 8. Methods of studying microorganisms. Microbiology is a science, which study most shallow living creatures - microorganisms. Before inventing of microscope humanity was in dark about their existence. But during the centuries people could make use of processes vital activity of microbes for its needs. They could prepare a koumiss, alcohol, wine, vinegar, bread, and other products. During many centuries the nature of fermentations remained incomprehensible. Microbiology learns morphology, physiology, genetics and microorganisms systematization, their ecology and the other life forms. Specific Classes of Microorganisms Algae Protozoa Fungi (yeasts and molds) Bacteria Rickettsiae Viruses Prions The Microorganisms are extraordinarily widely spread in nature. They literally ubiquitous forward us from birth to our death. Daily, hourly we eat up thousands and thousands of microbes together with air, water, food. -

The Effect of Tillage System and Crop Rotation on Soil Microbial Diversity and Composition in a Subtropical Acrisol
Diversity 2012, 4, 375-395; doi:10.3390/d4040375 OPEN ACCESS diversity ISSN 1424-2818 www.mdpi.com/journal/diversity Article The Effect of Tillage System and Crop Rotation on Soil Microbial Diversity and Composition in a Subtropical Acrisol Patricia Dorr de Quadros 1,2,*, Kateryna Zhalnina 2, Austin Davis-Richardson 2, Jennie R. Fagen 2, Jennifer Drew 2, Cimelio Bayer 1, Flavio A.O. Camargo 1 and Eric W. Triplett 2 1 Department of Soil Science, Federal University of Rio Grande do Sul, Porto Alegre, Brazil; E-Mails: [email protected] (C.B.); [email protected] (F.A.O.C.) 2 Department of Microbiology and Cell Science, Institute of Food and Agricultural Sciences, University of Florida, Gainesville, FL, USA; E-Mails: [email protected] (K.Z.), [email protected] (A.C.R.), [email protected] (J.D.), [email protected] (E.W.T.) *Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: (352)-575-5731. Received: 29 August 2012; in revised form: 11 October 2012 / Accepted: 19 October 2012 / Published: 31 October 2012 Abstract: Agricultural management alters physical and chemical soil properties, which directly affects microbial life strategies and community composition. The microbial community drives important nutrient cycling processes that can influence soil quality, cropping productivity and environmental sustainability. In this research, a long-term agricultural experiment in a subtropical Acrisol was studied in south Brazil. The plots at this site represent two tillage systems, two nitrogen fertilization regimes and three crop rotation systems. Using Illumina high-throughput sequencing of the 16S rRNA gene, the archaeal and bacterial composition was determined from phylum to species level in the different plot treatments. -

The Contribution of Syntrophic Fatty-Acid Degrading Microbial Communities to Anaerobic Digester Function and Stability
Marquette University e-Publications@Marquette Dissertations (2009 -) Dissertations, Theses, and Professional Projects The onC tribution of Syntrophic Fatty-Acid Degrading Microbial Communities to Anaerobic Digester Function and Stability Prince Peter Mathai Marquette University Recommended Citation Mathai, Prince Peter, "The onC tribution of Syntrophic Fatty-Acid Degrading Microbial Communities to Anaerobic Digester Function and Stability" (2015). Dissertations (2009 -). 653. https://epublications.marquette.edu/dissertations_mu/653 THE CONTRIBUTION OF SYNTROPHIC FATTY-ACID DEGRADING MICROBIAL COMMUNITIES TO ANAEROBIC DIGESTER FUNCTION AND STABILITY by Prince Peter Mathai, B.Tech. A Dissertation submitted to the Faculty of the Graduate School, Marquette University, in Partial Fulfillment of the Requirement for the Degree of Doctor of Philosophy Milwaukee, Wisconsin December 2015 ABSTRACT THE CONTRIBUTION OF SYNTROPHIC FATTY-ACID DEGRADING MICROBIAL COMMUNITIES TO ANAEROBIC DIGESTER FUNCTION AND STABILITY Prince Peter Mathai, B.Tech. Marquette University, 2015 Anaerobic digestion (AD), the conversion of complex organic matter to methane, occurs through a series of reactions mediated by different guilds of microorganisms. AD process imbalances, such as organic overload or high organic loading rates (OLR), can result in the accumulation of volatile fatty acids (VFA) e.g., propionate, which must be degraded to maintain stable reactor function. VFAs are metabolized by syntrophic fatty-acid degrading bacteria (SFAB) in association with methanogenic archaea (collectively, syntrophic microbial communities, SMC). Despite their indispensable role in AD, little is known about the ecology of SFAB, especially under stressed conditions. To facilitate ecological studies, four quantitative PCR assays, targeting propionate- and butyrate-degraders were developed, and applied to a variety of methanogenic environments. The highest SFAB abundance was observed in propionate enrichment cultures and anaerobic reactors.