Tree-Aggregated Predictive Modeling of Microbiome Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Marine Microbiome As Source of Natural Products
bs_bs_banner doi:10.1111/1751-7915.12882 Editorial: The microbiome as a source of new enterprises and job creation Marine microbiome as source of natural products Fernando de la Calle Department of Microbiology R&D, Pharma Mar S.A., Avda. de los Reyes, 1. Colmenar Viejo, 28770 Madrid, Spain. Less than 1% of living microorganisms can be cultured bioenergy and algal cosmetics and underestimate the in the laboratory, but even this minute part has produced role of the marine microbiome. Market opportunities and incredible discoveries such as antibiotics that have job creation are likely to significantly increase in the saved millions of lifes. We can only imagine what other future with the advent of transversal technologies direc- great inventions will come to light when the rest of this ted at exploiting other biotechnological advances, such enormous universe of living genomes is awakened. as the evolution of bioinformatics, synthetic biology, Everything is recorded in the genes, and modern molecular diagnostics and devices, biocatalysis and the metagenomic studies, involving the large-scale sequenc- many OMICS technologies. The marine microbiome ing of the genetic material of marine microorganisms, as must be an essential part of the bioeconomy. However, well as the similarities between the human gut and whilst the enormous potential of the marine microbiome ocean microbiomes, are not only providing powerful has been recognized by industry, there is currently a insights into why the Earth is a living planet, but are also lack of coordination amongst policy makers, govern- revealing possible causes and promising cures for meta- ments, civil society organizations, academia and large bolic diseases, including cancer. -

Diatoms Shape the Biogeography of Heterotrophic Prokaryotes in Early Spring in the Southern Ocean
Diatoms shape the biogeography of heterotrophic prokaryotes in early spring in the Southern Ocean Yan Liu, Pavla Debeljak, Mathieu Rembauville, Stéphane Blain, Ingrid Obernosterer To cite this version: Yan Liu, Pavla Debeljak, Mathieu Rembauville, Stéphane Blain, Ingrid Obernosterer. Diatoms shape the biogeography of heterotrophic prokaryotes in early spring in the Southern Ocean. Environmental Microbiology, Society for Applied Microbiology and Wiley-Blackwell, 2019, 21 (4), pp.1452-1465. 10.1111/1462-2920.14579. hal-02383818 HAL Id: hal-02383818 https://hal.archives-ouvertes.fr/hal-02383818 Submitted on 28 Nov 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Diatoms shape the biogeography of heterotrophic prokaryotes in early spring in the Southern Ocean 5 Yan Liu1, Pavla Debeljak1,2, Mathieu Rembauville1, Stéphane Blain1, Ingrid Obernosterer1* 1 Sorbonne Université, CNRS, Laboratoire d'Océanographie Microbienne, LOMIC, F-66650 10 Banyuls-sur-Mer, France 2 Department of Limnology and Bio-Oceanography, University of Vienna, A-1090 Vienna, Austria -

Novel Bacterial Lineages Associated with Boreal Moss Species Hannah
bioRxiv preprint doi: https://doi.org/10.1101/219659; this version posted November 16, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Novel bacterial lineages associated with boreal moss species 2 Hannah Holland-Moritz1,2*, Julia Stuart3, Lily R. Lewis4, Samantha Miller3, Michelle C. Mack3, Stuart 3 F. McDaniel4, Noah Fierer1,2* 4 Affiliations: 5 1Cooperative Institute for Research in Environmental Sciences, University of Colorado at Boulder, 6 Boulder, CO, USA 7 2Department of Ecology and Evolutionary Biology, University of Colorado at Boulder, Boulder, CO, 8 USA 9 3Center for Ecosystem Science and Society, Northern Arizona University, Flagstaff, AZ USA 10 4Department of Biology, University of Florida, Gainesville, FL 32611-8525, USA 11 *Corresponding Author 12 13 Abstract 14 Mosses are critical components of boreal ecosystems where they typically account for a large 15 proportion of net primary productivity and harbor diverse bacterial communities that can be the major 16 source of biologically-fixed nitrogen in these ecosystems. Despite their ecological importance, we have 17 limited understanding of how microbial communities vary across boreal moss species and the extent to 18 which local environmental conditions may influence the composition of these bacterial communities. 19 We used marker gene sequencing to analyze bacterial communities associated with eight boreal moss 20 species collected near Fairbanks, AK USA. We found that host identity was more important than site in 21 determining bacterial community composition and that mosses harbor diverse lineages of potential N2- 22 fixers as well as an abundance of novel taxa assigned to understudied bacterial phyla (including 23 candidate phylum WPS-2). -

Genomic Analysis of Family UBA6911 (Group 18 Acidobacteria)
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.09.439258; this version posted April 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 2 Genomic analysis of family UBA6911 (Group 18 3 Acidobacteria) expands the metabolic capacities of the 4 phylum and highlights adaptations to terrestrial habitats. 5 6 Archana Yadav1, Jenna C. Borrelli1, Mostafa S. Elshahed1, and Noha H. Youssef1* 7 8 1Department of Microbiology and Molecular Genetics, Oklahoma State University, Stillwater, 9 OK 10 *Correspondence: Noha H. Youssef: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2021.04.09.439258; this version posted April 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 11 Abstract 12 Approaches for recovering and analyzing genomes belonging to novel, hitherto unexplored 13 bacterial lineages have provided invaluable insights into the metabolic capabilities and 14 ecological roles of yet-uncultured taxa. The phylum Acidobacteria is one of the most prevalent 15 and ecologically successful lineages on earth yet, currently, multiple lineages within this phylum 16 remain unexplored. Here, we utilize genomes recovered from Zodletone spring, an anaerobic 17 sulfide and sulfur-rich spring in southwestern Oklahoma, as well as from multiple disparate soil 18 and non-soil habitats, to examine the metabolic capabilities and ecological role of members of 19 the family UBA6911 (group18) Acidobacteria. -

Major Ocean Currents May Shape the Microbiome of the Topshell Phorcus
www.nature.com/scientificreports OPEN Major ocean currents may shape the microbiome of the topshell Phorcus sauciatus in the NE Atlantic Ocean Ricardo Sousa1,2,3, Joana Vasconcelos3,4,5, Iván Vera‑Escalona5, João Delgado2,6, Mafalda Freitas1,2,3, José A. González7 & Rodrigo Riera5,8* Studies on microbial communities are pivotal to understand the role and the evolutionary paths of the host and their associated microorganisms in the ecosystems. Meta‑genomics techniques have proven to be one of the most efective tools in the identifcation of endosymbiotic communities of host species. The microbiome of the highly exploited topshell Phorcus sauciatus was characterized in the Northeastern Atlantic (Portugal, Madeira, Selvagens, Canaries and Azores). Alpha diversity analysis based on observed OTUs showed signifcant diferences among regions. The Principal Coordinates Analysis of beta‑diversity based on presence/absence showed three well diferentiated groups, one from Azores, a second from Madeira and the third one for mainland Portugal, Selvagens and the Canaries. The microbiome results may be mainly explained by large‑scale oceanographic processes of the study region, i.e., the North Atlantic Subtropical Gyre, and specifcally by the Canary Current. Our results suggest the feasibility of microbiome as a model study to unravel biogeographic and evolutionary processes in marine species with high dispersive potential. During the last decades we have observed an increase in the number of studies trying to elucidate the role of spe- cies and the environment where they live due to research expeditions and the use of several modern techniques to identify species, including genetic-based techniques 1. Early studies based on genetics focused on the description of species and populations but soon afer the frst results, it was evident that genetic-based studies could also be used to identify and describe major biogeographic patterns as well as to create the pathway to evaluate new hypotheses and ecological questions 2–4. -

Corals and Sponges Under the Light of the Holobiont Concept: How Microbiomes Underpin Our Understanding of Marine Ecosystems
fmars-08-698853 August 11, 2021 Time: 11:16 # 1 REVIEW published: 16 August 2021 doi: 10.3389/fmars.2021.698853 Corals and Sponges Under the Light of the Holobiont Concept: How Microbiomes Underpin Our Understanding of Marine Ecosystems Chloé Stévenne*†, Maud Micha*†, Jean-Christophe Plumier and Stéphane Roberty InBioS – Animal Physiology and Ecophysiology, Department of Biology, Ecology & Evolution, University of Liège, Liège, Belgium In the past 20 years, a new concept has slowly emerged and expanded to various domains of marine biology research: the holobiont. A holobiont describes the consortium formed by a eukaryotic host and its associated microorganisms including Edited by: bacteria, archaea, protists, microalgae, fungi, and viruses. From coral reefs to the Viola Liebich, deep-sea, symbiotic relationships and host–microbiome interactions are omnipresent Bremen Society for Natural Sciences, and central to the health of marine ecosystems. Studying marine organisms under Germany the light of the holobiont is a new paradigm that impacts many aspects of marine Reviewed by: Carlotta Nonnis Marzano, sciences. This approach is an innovative way of understanding the complex functioning University of Bari Aldo Moro, Italy of marine organisms, their evolution, their ecological roles within their ecosystems, and Maria Pia Miglietta, Texas A&M University at Galveston, their adaptation to face environmental changes. This review offers a broad insight into United States key concepts of holobiont studies and into the current knowledge of marine model *Correspondence: holobionts. Firstly, the history of the holobiont concept and the expansion of its use Chloé Stévenne from evolutionary sciences to other fields of marine biology will be discussed. -

A Community Perspective on the Concept of Marine Holobionts: Current Status, Challenges, and Future Directions
A community perspective on the concept of marine holobionts: current status, challenges, and future directions Simon M. Dittami1, Enrique Arboleda2, Jean-Christophe Auguet3, Arite Bigalke4, Enora Briand5, Paco Cárdenas6, Ulisse Cardini7, Johan Decelle8, Aschwin H. Engelen9, Damien Eveillard10, Claire M.M. Gachon11, Sarah M. Griffiths12, Tilmann Harder13, Ehsan Kayal2, Elena Kazamia14, Francois¸ H. Lallier15, Mónica Medina16, Ezequiel M. Marzinelli17,18,19, Teresa Maria Morganti20, Laura Núñez Pons21, Soizic Prado22, José Pintado23, Mahasweta Saha24,25, Marc-André Selosse26,27, Derek Skillings28, Willem Stock29, Shinichi Sunagawa30, Eve Toulza31, Alexey Vorobev32, Catherine Leblanc1 and Fabrice Not15 1 Integrative Biology of Marine Models (LBI2M), Station Biologique de Roscoff, Sorbonne Université, CNRS, Roscoff, France 2 FR2424, Station Biologique de Roscoff, Sorbonne Université, CNRS, Roscoff, France 3 MARBEC, Université de Montpellier, CNRS, IFREMER, IRD, Montpellier, France 4 Institute for Inorganic and Analytical Chemistry, Bioorganic Analytics, Friedrich-Schiller-Universität Jena, Jena, Germany 5 Laboratoire Phycotoxines, Ifremer, Nantes, France 6 Pharmacognosy, Department of Medicinal Chemistry, Uppsala University, Uppsala, Sweden 7 Integrative Marine Ecology Dept, Stazione Zoologica Anton Dohrn, Napoli, Italy 8 Laboratoire de Physiologie Cellulaire et Végétale, Université Grenoble Alpes, CNRS, CEA, INRA, Grenoble, France 9 CCMAR, Universidade do Algarve, Faro, Portugal 10 Laboratoire des Sciences Numériques de Nantes (LS2N), Université -

The Marine Microbiome Initiative
The Marine Microbiome Initiative Justin Seymour, Martin Ostrowski, Mark Brown, Lev Bodrossy, Jodie van de Kamp, Andrew Bissett, Ana Lara-Lopez Seymour 2014 Emerging EOV: Microbial diversity and biomass Slides from Pier Buttigieg (via Ana Lara-Lopez) Evolution of the Marine Microbiome Initiative 2012 Australian Marine Microbe Biodiversity Initiative (AMMBI) NSI PHB MAI Evolution of the Marine Microbiome Initiative 2012 2014 Australian Marine Microbe BPA Marine Microbes Project Biodiversity Initiative (AMMBI) $1M DAR YON NSI NSI ROT PHB PHB KAI MAI MAI Evolution of the Marine Microbiome Initiative 2012 2014 2018 Australian Marine Microbe BPA Marine Microbes Project Biodiversity Initiative (AMMBI) Marine Microbes Project + Biomes of Australian DAR Soil Environments YON NSI NSI ROT PHB PHB KAI MAI MAI Evolution of the Marine Microbiome Initiative 2012 2014 2018 Australian Marine Microbe BPA Marine Microbes Project Biodiversity Initiative (AMMBI) DAR 2019 YON Marine Microbiome Initiative A new IMOS Facility! NSI NSI ROT • Sample processing & PHB PHB archiving KAI • DNA extractions MAI MAI Jodie van de Kamp Consortium of > 50 researchers from 10 universities and research institutes Contributed $910K DNA extraction: $90,000 (Marine Microbes Init. Facility) Bioinformatic position: $560,000 (Bioinformatics Sub-Facility) Coastal microbial observatory support: $260,000 Andrew Bissett Bioinformatics New IMOS Sub-Facility! • Genomics data processing Matt Smith • Workflows NSI YON The Australian DAR Microbiome dataset Dark Ocean PHB contains ~5,000 -

Microbial Communities of Polymetallic Deposits' Acidic
Microbial Communities of Polymetallic Deposits’ Acidic Ecosystems of ANGOR UNIVERSITY Continental Climatic Zone With High Temperature Contrasts Gavrilov, Sergei N.; Korzhenkov, Aleksei A.; Kublanov, Ilya V.; Bargiela, Rafael; Zamana, Leonid V.; Toshchakov, Stepan V.; Golyshin, Peter; Golyshina, Olga Frontiers in Microbiology PRIFYSGOL BANGOR / B Published: 17/07/2019 Publisher's PDF, also known as Version of record Cyswllt i'r cyhoeddiad / Link to publication Dyfyniad o'r fersiwn a gyhoeddwyd / Citation for published version (APA): Gavrilov, S. N., Korzhenkov, A. A., Kublanov, I. V., Bargiela, R., Zamana, L. V., Toshchakov, S. V., Golyshin, P., & Golyshina, O. (2019). Microbial Communities of Polymetallic Deposits’ Acidic Ecosystems of Continental Climatic Zone With High Temperature Contrasts. Frontiers in Microbiology. Hawliau Cyffredinol / General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. 01. Oct. 2021 ORIGINAL RESEARCH published: 17 July 2019 doi: 10.3389/fmicb.2019.01573 Microbial Communities of Polymetallic Deposits’ Acidic Ecosystems of Continental Climatic Zone With High Temperature Contrasts Sergey N. -

Phylogenetic Responses of Marine Free-Living Bacterial Community to Phaeocystis Globosa Bloom in Beibu Gulf, China
fmicb-11-01624 July 14, 2020 Time: 17:42 # 1 ORIGINAL RESEARCH published: 16 July 2020 doi: 10.3389/fmicb.2020.01624 Phylogenetic Responses of Marine Free-Living Bacterial Community to Phaeocystis globosa Bloom in Beibu Gulf, China Nan Li1*, Huaxian Zhao1, Gonglingxia Jiang1, Qiangsheng Xu1, Jinli Tang1, Xiaoli Li1, Jiemei Wen1, Huimin Liu1, Chaowu Tang1, Ke Dong2 and Zhenjun Kang3* 1 Key Laboratory of Environment Change and Resources Use in Beibu Gulf, Ministry of Education, Nanning Normal University, Nanning, China, 2 Department of Biological Sciences, Kyonggi University, Suwon-si, South Korea, 3 Guangxi Key Laboratory of Marine Disaster in the Beibu Gulf, Beibu Gulf University, Qinzhou, China Phaeocystis globosa blooms are recognized as playing an essential role in shaping the structure of the marine community and its functions in marine ecosystems. In this study, we observed variation in the alpha diversity and composition of marine free-living bacteria during P. globosa blooms and identified key microbial community Edited by: assembly patterns during the blooms. The results showed that the Shannon index Olga Lage, was higher before the blooming of P. globosa in the subtropical bay. Marinobacterium University of Porto, Portugal (g-proteobacteria), Erythrobacter (a-proteobacteria), and Persicobacter (Cytophagales) Reviewed by: Xiaoqian Yu, were defined as the most important genera, and they were more correlated with University of Vienna, Austria environmental factors at the terminal stage of P. globosa blooms. Furthermore, different Catarina Magalhães, community assembly processes were observed. Both the mean nearest relatedness University of Porto, Portugal index (NRI) and nearest taxon index (NTI) revealed the dominance of deterministic *Correspondence: Nan Li factors in the non-blooming and blooming periods of P. -
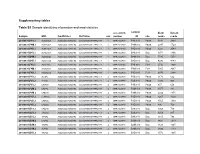
Supplementary Tables Table S1 Sample Identifying Information And
Supplementary tables Table S1 Sample identifying information and read statistics accession sample #raw #clean Sample MID FwdPrimer RvPrimer run number ID site reads reads 20100831.P4S.1 ACACGAGA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 P4S.910 Palsa 3831 2893 20100831.P4M.1 ACATAGCA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 P4M.910 Palsa 2047 1729 20100831.P4D.1 ACGATACA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 P4D.910 Palsa 3221 2543 20100831.B4S.1 ACTGCTCA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 B4S.910 Bog 1971 1366 20100831.B4M.1 AGCAGCGC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 B4M.910 Bog 5530 5251 20100831.B4D.1 AGCGTGCA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 B4D.910 Bog 4296 4143 20100831.F4S.1 AGTCTAGC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 F4S.910 Fen* 2716 1890 20100831.F4M.1 ATGACTGA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 F4M.910 Fen* 5005 3987 20100831.F4D.1 ATGCACGC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 F4D.910 Fen* 2870 2384 20100901.P1S.2 ACTGAT ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P1S.910 Palsa 1170 662 20100901.P2S.2 ATGTGT ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P2S.910 Palsa 1320 923 20100901.P3S.2 CACAGT ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P3S.910 Palsa 857 656 20100901.P2M.2 CACTAC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P2M.910 Palsa 1077 881 20100901.P3M.2 CAGCAT ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P3M.910 Palsa 2032 1474 20100901.P1D.2 CTAGAGC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT -

Pyrosequencing Investigation Into the Bacterial Community in Permafrost Soils Along the China-Russia Crude Oil Pipeline (CRCOP)
Pyrosequencing Investigation into the Bacterial Community in Permafrost Soils along the China-Russia Crude Oil Pipeline (CRCOP) Sizhong Yang1*, Xi Wen2, Huijun Jin1, Qingbai Wu1 1 State Key Laboratory of Frozen Soil Engineering (SKLFSE), Cold and Arid Regions Environmental and Engineering Research Institute (CAREERI), Chinese Academy of Sciences, Lanzhou, China, 2 College of Electrical Engineering, Northwest University for Nationalities, Lanzhou, China Abstract The China-Russia Crude Oil Pipeline (CRCOP) goes through 441 km permafrost soils in northeastern China. The bioremediation in case of oil spills is a major concern. So far, little is known about the indigenous bacteria inhabiting in the permafrost soils along the pipeline. A pilot 454 pyrosequencing analysis on the communities from four selected sites which possess high environment risk along the CRCOP is herein presented. The results reveal an immense bacterial diversity than previously anticipated. A total of 14448 OTUs with 84834 reads are identified, which could be assigned into 39 different phyla, and 223 families or 386 genera. Only five phyla sustain a mean OTU abundance more than 5% in all the samples, but they altogether account for 85.08% of total reads. Proteobacteria accounts for 41.65% of the total OTUs or 45% of the reads across all samples, and its proportion generally increases with soil depth, but OTUs numerically decline. Among Proteobacteria, the abundance of Beta-, Alpha-, Delta- and Gamma- subdivisions average to 38.7% (2331 OTUs), 37.5% (2257 OTUs), 10.35% (616 OTUs), and 6.21% (374 OTUs), respectively. Acidobacteria (esp. Acidobacteriaceae), Actinobacteria (esp. Intrasporangiaceae), Bacteroidetes (esp. Sphingobacteria and Flavobacteria) and Chloroflexi (esp.