Hyperactive Alpha2-Chimaerin Reveals the Complexity of Axon Guidance Signaling Pathways in Motor Neuron Development
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Regulation of the Mammalian Target of Rapamycin Complex 2 (Mtorc2)
Regulation of the Mammalian Target Of Rapamycin Complex 2 (mTORC2) Inauguraldissertation Zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Klaus-Dieter Molle aus Heilbronn, Deutschland Basel, 2006 Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät Auf Antrag von Prof. Michael N. Hall und Prof. Markus Affolter. Basel, den 21.11.2006 Prof. Hans-Peter Hauri Dekan Summary The growth controlling mammalian Target of Rapamycin (mTOR) is a conserved Ser/Thr kinase found in two structurally and functionally distinct complexes, mTORC1 and mTORC2. The tumor suppressor TSC1-TSC2 complex inhibits mTORC1 by acting on the small GTPase Rheb, but the role of TSC1-TSC2 and Rheb in the regulation of mTORC2 is unclear. Here we examined the role of TSC1-TSC2 in the regulation of mTORC2 in human embryonic kidney 293 cells. Induced knockdown of TSC1 and TSC2 (TSC1/2) stimulated mTORC2-dependent actin cytoskeleton organization and Paxillin phosphorylation. Furthermore, TSC1/2 siRNA increased mTORC2-dependent Ser473 phosphorylation of plasma membrane bound, myristoylated Akt/PKB. This suggests that loss of Akt/PKB Ser473 phosphorylation in TSC mutant cells, as reported previously, is due to inhibition of Akt/PKB localization rather than inhibition of mTORC2 activity. Amino acids and overexpression of Rheb failed to stimulate mTORC2 signaling. Thus, TSC1-TSC2 also inhibits mTORC2, but possibly independently of Rheb. Our results suggest that mTORC2 hyperactivation may contribute to the pathophysiology of diseases such as cancer and Tuberous Sclerosis Complex. i Acknowledgement During my PhD studies in the Biozentrum I received a lot of support from many people around me who I mention here to express my gratefulness. -

Lymphocyte/WBC-Based Comprehensive Testing Via Next-Gen Sequencing NF1/SPRED1 and Other Rasopathy Related Conditions on Blood/Saliva
Last Updated October 2019 Institutional Price TAT Genetic Test CPT Codes Z codes Specimen Requirements (USD$) (working days) Lymphocyte/WBC-based Comprehensive Testing via Next-Gen Sequencing NF1/SPRED1 and Other RASopathy Related Conditions on Blood/Saliva NF1- only NGS testing and copy number analysis for the NF1 gene (NF1-NG) This testing includes an average coverage of >1600x to allow for the identification of (1) 3-6ml whole blood in EDTA (purple topped) tubes mosaicism as low as 3-5% of the alleles. In addition, novel variants identified in the $1,000 81408 25 (2) Oragene 575 saliva kit (provided by the MGL) ZB6A9 NF1 gene will be confirmed via RNA-based analysis at no additional charge. RNA- $1,600 (RUSH) 81479 15 (RUSH) (3) DNA sample (25ul volume at 3ug, O.D. value at based testing will also be provided to non-founder, multigenerational families with 260:280 ≥1.8) “classic” NF1 at no additional charge if next-generation sequencing is found negative. (1) 3-6ml whole blood in EDTA (purple topped) tubes SPRED1-only NGS testing and copy number analysis for SPRED1 (SPD1-NG) $800 81405 25 (2) Oragene 575 saliva kit (provided by the MGL) This testing includes an average coverage of >1600x to allow for the identification of ZB6AC $1,400 (RUSH) 81479 15 (RUSH) (3) DNA sample (25ul volume at 3ug, O.D. value at mosaicism as low as 3-5% of the alleles after comprehensive NF1 analysis. 260:280 ≥1.8) NF1/SPRED1 NGS testing and copy number analysis for NF1 and SPRED1 (NFSP-NG) (1) 3-6ml whole blood in EDTA (purple topped) tubes 81408 This testing includes an average coverage of >1600x to allow for the identification of $1,100 25 (2) Oragene 575 saliva kit (provided by the MGL) 81405 ZB6A8 mosaicism as low as 3-5% of the alleles. -

Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association Between BMI and Adult-Onset Non- Atopic
Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association between BMI and Adult-Onset Non- Atopic Asthma Ayoung Jeong 1,2, Medea Imboden 1,2, Akram Ghantous 3, Alexei Novoloaca 3, Anne-Elie Carsin 4,5,6, Manolis Kogevinas 4,5,6, Christian Schindler 1,2, Gianfranco Lovison 7, Zdenko Herceg 3, Cyrille Cuenin 3, Roel Vermeulen 8, Deborah Jarvis 9, André F. S. Amaral 9, Florian Kronenberg 10, Paolo Vineis 11,12 and Nicole Probst-Hensch 1,2,* 1 Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; [email protected] (A.J.); [email protected] (M.I.); [email protected] (C.S.) 2 Department of Public Health, University of Basel, 4001 Basel, Switzerland 3 International Agency for Research on Cancer, 69372 Lyon, France; [email protected] (A.G.); [email protected] (A.N.); [email protected] (Z.H.); [email protected] (C.C.) 4 ISGlobal, Barcelona Institute for Global Health, 08003 Barcelona, Spain; [email protected] (A.-E.C.); [email protected] (M.K.) 5 Universitat Pompeu Fabra (UPF), 08002 Barcelona, Spain 6 CIBER Epidemiología y Salud Pública (CIBERESP), 08005 Barcelona, Spain 7 Department of Economics, Business and Statistics, University of Palermo, 90128 Palermo, Italy; [email protected] 8 Environmental Epidemiology Division, Utrecht University, Institute for Risk Assessment Sciences, 3584CM Utrecht, Netherlands; [email protected] 9 Population Health and Occupational Disease, National Heart and Lung Institute, Imperial College, SW3 6LR London, UK; [email protected] (D.J.); [email protected] (A.F.S.A.) 10 Division of Genetic Epidemiology, Medical University of Innsbruck, 6020 Innsbruck, Austria; [email protected] 11 MRC-PHE Centre for Environment and Health, School of Public Health, Imperial College London, W2 1PG London, UK; [email protected] 12 Italian Institute for Genomic Medicine (IIGM), 10126 Turin, Italy * Correspondence: [email protected]; Tel.: +41-61-284-8378 Int. -

Tuberous Sclerosis Complex: Molecular Pathogenesis and Animal Models
Neurosurg Focus 20 (1):E4, 2006 Tuberous sclerosis complex: molecular pathogenesis and animal models LEANDRO R. PIEDIMONTE, M.D., IAN K. WAILES, M.D., AND HOWARD L. WEINER, M.D. Division of Pediatric Neurosurgery, Department of Neurosurgery, New York University School of Medicine, New York, New York Mutations in one of two genes, TSC1 and TSC2, result in a similar disease phenotype by disrupting the normal inter- action of their protein products, hamartin and tuberin, which form a functional signaling complex. Disruption of these genes in the brain results in abnormal cellular differentiation, migration, and proliferation, giving rise to the charac- teristic brain lesions of tuberous sclerosis complex (TSC) called cortical tubers. The most devastating complications of TSC affect the central nervous system and include epilepsy, mental retardation, autism, and glial tumors. Relevant animal models, including conventional and conditional knockout mice, are valuable tools for studying the normal func- tions of tuberin and hamartin and the way in which disruption of their expression gives rise to the variety of clinical features that characterize TSC. In the future, these animals will be invaluable preclinical models for the development of highly specific and efficacious treatments for children affected with TSC. KEY WORDS • tuberous sclerosis • pathogenesis • animal model Tuberous sclerosis complex is an autosomal-dominant zures, mental retardation, and autism.1,51 The number and tumor predisposition syndrome that affects approximately location of cortical tubers and the patient’s age at seizure 1 in 7500 individuals worldwide.19 The TSC is character- onset are highly correlated with neurological outcomes.1,30 ized by benign hamartomatous growths in multiple organs, Epilepsy occurs in approximately 80% of affected indi- including the kidney, skin, retina, lung, and brain. -

Supplementary Table 2
Supplementary Table 2. Differentially Expressed Genes following Sham treatment relative to Untreated Controls Fold Change Accession Name Symbol 3 h 12 h NM_013121 CD28 antigen Cd28 12.82 BG665360 FMS-like tyrosine kinase 1 Flt1 9.63 NM_012701 Adrenergic receptor, beta 1 Adrb1 8.24 0.46 U20796 Nuclear receptor subfamily 1, group D, member 2 Nr1d2 7.22 NM_017116 Calpain 2 Capn2 6.41 BE097282 Guanine nucleotide binding protein, alpha 12 Gna12 6.21 NM_053328 Basic helix-loop-helix domain containing, class B2 Bhlhb2 5.79 NM_053831 Guanylate cyclase 2f Gucy2f 5.71 AW251703 Tumor necrosis factor receptor superfamily, member 12a Tnfrsf12a 5.57 NM_021691 Twist homolog 2 (Drosophila) Twist2 5.42 NM_133550 Fc receptor, IgE, low affinity II, alpha polypeptide Fcer2a 4.93 NM_031120 Signal sequence receptor, gamma Ssr3 4.84 NM_053544 Secreted frizzled-related protein 4 Sfrp4 4.73 NM_053910 Pleckstrin homology, Sec7 and coiled/coil domains 1 Pscd1 4.69 BE113233 Suppressor of cytokine signaling 2 Socs2 4.68 NM_053949 Potassium voltage-gated channel, subfamily H (eag- Kcnh2 4.60 related), member 2 NM_017305 Glutamate cysteine ligase, modifier subunit Gclm 4.59 NM_017309 Protein phospatase 3, regulatory subunit B, alpha Ppp3r1 4.54 isoform,type 1 NM_012765 5-hydroxytryptamine (serotonin) receptor 2C Htr2c 4.46 NM_017218 V-erb-b2 erythroblastic leukemia viral oncogene homolog Erbb3 4.42 3 (avian) AW918369 Zinc finger protein 191 Zfp191 4.38 NM_031034 Guanine nucleotide binding protein, alpha 12 Gna12 4.38 NM_017020 Interleukin 6 receptor Il6r 4.37 AJ002942 -

Neurofibromatosis-1 Regulates Mtor-Mediated Astrocyte Growth
Neurofibromatosis-1 regulates mTOR-mediated astrocyte growth and glioma formation in a TSC/Rheb-independent manner Sutapa Banerjee, Nikkilina R. Crouse, Ryan J. Emnett, Scott M. Gianino, and David H. Gutmann1 Department of Neurology, Washington University School of Medicine, St. Louis, MO 63110 Edited* by Webster K. Cavenee, Ludwig Institute, University of California at San Diego, La Jolla, CA, and approved August 15, 2011 (received for review December 17, 2010) Converging evidence from the analysis of human brain tumors seizures and death, whereas Nf1 loss in these same neuroglial and genetically engineered mice has revealed that the mamma- progenitors has no effect on mouse viability or epileptogenesis (16, lian target of rapamycin (mTOR) pathway is a central regulator of 17). Importantly, although Pten-, hamartin-, and neurofibromin- glial and glioma cell growth. In this regard, mutational inactiva- deficient glial phenotypes have different biological effects, each tion of neurofibromatosis-1 (NF1), tuberous sclerosis complex is completely inhibited by rapamycin in vitro and in vivo (15, 18– (TSC), and PTEN genes is associated with glioma formation, such 21). Collectively, these observations support a model in which fi that pharmacologic inhibition of mTOR signaling results in atten- neuro bromin-regulated mTOR-dependent growth regulation uated tumor growth. This shared dependence on mTOR suggests is distinct from TSC/Rheb-mediated mTOR-dependent growth that PTEN and NF1 (neurofibromin) glial growth regulation regulation in the brain. To -

Snapshot: Mtorc1 Signaling at the Lysosomal Surface Liron Bar-Peled and David M
1390 Cell SnapShot: mTORC1 Signaling at the 151 , December 7,2012©2012 ElsevierInc. DOI http://dx.doi.org/10.1016/j.cell.2012.11.038 Lysosomal Surface Liron Bar-Peled and David M. Sabatini Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology, Cambridge, MA 02142, USA Nutrient signaling Wnt Growth Leu signaling factor signaling TNF signaling Amino Gln Wnt Tyrosine acids Frizzled IGF kinase TNFD receptor TNF receptor DNA Energy levels O levels SLC1A5 SLC7A5 2 damage ATP/AMP PIP2 Pten Amino acid mTORC1 Dsh1 IRS1 transporter Gln (inactive) GRB2 SOS PI3K PIP3 Gln Redd1 p53 LKB1 GEF GSK3 activity Leu GTP Ras NF1 PDK1 GAP Sestrin Movement to the activity RagAGTP lysosomal surface Movement AMPK Raf GDP away from Rapamycin RagA the lysosomal surface FKBP12 Mek Erk1/2 Rsk1 Akt1 IKK` v-ATPase GEF activity CYTOPLASM RagAGTP mTORC1 Ragulator GTP GAP activity GDP TSC complex RagC (active) Rheb Tumor suppressor Oncogene mTORC1 substrate ? Growth DOWNSTREAM CELLULAR PROGRAMS REGULATED BY mTORC1 ACTIVITY See online version for legend and references. S6K1 Amino acids LYSOSOME Protein synthesis 4EBP1 COMPLEXES AT THE LYSOSOMAL SURFACE HIF1_ Energy metabolism A B A E E G G pras40 deptor Lysosome biogenesis A BB MP1 HBXIP TSC1 TBC1D7 TFEB p14 C7orf59 raptor H mLST8 Lipin-1 D C Lipid p18 biosynthesis a d F mTOR TSC2 SREBP1/2 c cc ATG13 FIP200 Autophagy Lysosomal v-ATPase Ragulator complex mTORC1 TSC complex ULK1 SnapShot: mTORC1 Signaling at the Lysosomal Surface Liron Bar-Peled and David M. Sabatini Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology, Cambridge, MA 02142, USA In mammals, the mTOR complex 1 (mTORC1) ser/thr kinase regulates cellular and organismal growth in response to a variety of environmental and intracellular stimuli. -

Supplemental Table 1
Supplemental Data Supplemental Table 1. Genes differentially regulated by Ad-KLF2 vs. Ad-GFP infected EC. Three independent genome-wide transcriptional profiling experiments were performed, and significantly regulated genes were identified. Color-coding scheme: Up, p < 1e-15 Up, 1e-15 < p < 5e-10 Up, 5e-10 < p < 5e-5 Up, 5e-5 < p <.05 Down, p < 1e-15 As determined by Zpool Down, 1e-15 < p < 5e-10 Down, 5e-10 < p < 5e-5 Down, 5e-5 < p <.05 p<.05 as determined by Iterative Standard Deviation Algorithm as described in Supplemental Methods Ratio RefSeq Number Gene Name 1,058.52 KRT13 - keratin 13 565.72 NM_007117.1 TRH - thyrotropin-releasing hormone 244.04 NM_001878.2 CRABP2 - cellular retinoic acid binding protein 2 118.90 NM_013279.1 C11orf9 - chromosome 11 open reading frame 9 109.68 NM_000517.3 HBA2;HBA1 - hemoglobin, alpha 2;hemoglobin, alpha 1 102.04 NM_001823.3 CKB - creatine kinase, brain 96.23 LYNX1 95.53 NM_002514.2 NOV - nephroblastoma overexpressed gene 75.82 CeleraFN113625 FLJ45224;PTGDS - FLJ45224 protein;prostaglandin D2 synthase 21kDa 74.73 NM_000954.5 (brain) 68.53 NM_205545.1 UNQ430 - RGTR430 66.89 NM_005980.2 S100P - S100 calcium binding protein P 64.39 NM_153370.1 PI16 - protease inhibitor 16 58.24 NM_031918.1 KLF16 - Kruppel-like factor 16 46.45 NM_024409.1 NPPC - natriuretic peptide precursor C 45.48 NM_032470.2 TNXB - tenascin XB 34.92 NM_001264.2 CDSN - corneodesmosin 33.86 NM_017671.3 C20orf42 - chromosome 20 open reading frame 42 33.76 NM_024829.3 FLJ22662 - hypothetical protein FLJ22662 32.10 NM_003283.3 TNNT1 - troponin T1, skeletal, slow LOC388888 (LOC388888), mRNA according to UniGene - potential 31.45 AK095686.1 CONFLICT - LOC388888 (na) according to LocusLink. -

Cardiovascular Diseases Genetic Testing Program Information
Cardiovascular Diseases Genetic Testing Program Description: Congenital Heart Disease Panels We offer comprehensive gene panels designed to • Congenital Heart Disease Panel (187 genes) diagnose the most common genetic causes of hereditary • Heterotaxy Panel (114 genes) cardiovascular diseases. Testing is available for congenital • RASopathy/Noonan Spectrum Disorders Panel heart malformation, cardiomyopathy, arrythmia, thoracic (31 genes) aortic aneurysm, pulmonary arterial hypertension, Marfan Other Panels syndrome, and RASopathy/Noonan spectrum disorders. • Pulmonary Arterial Hypertension (PAH) Panel Hereditary cardiovascular disease is caused by variants in (20 genes) many different genes, and may be inherited in an autosomal dominant, autosomal recessive, or X-linked manner. Other Indications: than condition-specific panels, we also offer single gene Panels: sequencing for any gene on the panels, targeted variant • Confirmation of genetic diagnosis in a patient with analysis, and targeted deletion/duplication analysis. a clinical diagnosis of cardiovascular disease Tests Offered: • Carrier or pre-symptomatic diagnosis identification Arrythmia Panels in individuals with a family history of cardiovascular • Comprehensive Arrhythmia Panel (81 genes) disease of unknown genetic basis • Atrial Fibrillation (A Fib) Panel (28 genes) Gene Specific Sequencing: • Atrioventricular Block (AV Block) Panel (7 genes) • Confirmation of genetic diagnosis in a patient with • Brugada Syndrome Panel (21 genes) cardiovascular disease and in whom a specific -
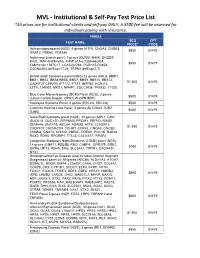
MVL - Institutional & Self-Pay Test Price List *All Prices Are for Institutional Clients and Self-Pay ONLY
MVL - Institutional & Self-Pay Test Price List *All prices are for institutional clients and self-pay ONLY. A $500 fee will be assessed for individuals paying with insurance. PANELS SEQ CPT TEST NAME PRICE* CODE Achromatopsiapanel (NGS), 6 genes (ATF6, CNGA3, CNGB3, $950 81479 GNAT2, PDE6C, PDE6H) Ashkenazi Jewish panel, 7 genes (CLRN1-N48K, DHDDS- K42E, MAK-K429insAlu, FAM161A-c.1355-6delCA, $550 81407 FAM161Ac.1567C>T, LCA5-Q279X, PCDH15-R245X, CACNA2D4-delExon17-26, TRPM1-delExon2-7) Bardet-Biedl Syndrome panel (NGS) 23 genes (ARL6, BBIP1, BBS1, BBS2, BBS4,BBS5, BBS7, BBS9, BBS10, BBS12, $1,500 81479 C8ORF37,CEP290, IFT172, IFT27, INPP5E, KCNJ13, LZTFL1,MKKS, MKS1, NPHP1, SDCCAG8, TRIM32, TTC8) Blue Cone Monochromacy (BCM) Panel (NGS), 2 genes $500 81479 (Locus Control Region, OPN1LW-OPN1MW) Cockayne Sydrome Panel, 2 genes (ERCC6, ERCC8) $500 81479 Common Hearing Loss Panel, 3 genes (SLC26A4, GJB2, $650 81479 GJB6) Cone-Rod Dystrophy panel (NGS), 33 genes (AIPL1, CRX, GUCA1A, GUCY2D, PITPNM3, PROM1, PRPH2, RIMS1, SEMA4A, UNC119, ABCA4, ADAM9, ATF6, C21ORF2, $1,950 81479 C8ORF37,CACNA2D4, CDHR1, CERKL, CNGA3, CNGB3, CNNM4, GNAT2, KCNV2, PDE6C, PDE6H, POC1B, RAB28, RAX2, RDH5, RPGRIP1, TTLL5, CACNA1F, RPGR) Congenital Stationary Night Blindness (CSNB) panel (NGS), 14 genes (GNAT1, PDE6B, RHO, CABP4, GPR179, GRK1, $950 81479 GRM6,LRIT3, RDH5, SAG, SLC24A1, TRPM1, CACNA1F, NYX) Developmental Eye Disease (also includes Anterior Segment Dysgenesis) panel v4: 59 genes (ABCB6, ALDH1A3, ATOH7, B3GALTL, BCOR, BMP4, c12orf57, CASK, CHD7, COL4A1, -

Ubiquitination of Rheb Governs Growth Factor-Induced Mtorc1 Activation
www.nature.com/cr www.cell-research.com ARTICLE OPEN Ubiquitination of Rheb governs growth factor-induced mTORC1 activation Lu Deng1, Lei Chen1,2,3, Linlin Zhao1, Yan Xu4, Xiaoping Peng1, Xinbo Wang1, Lin Ding1, Jiali Jin1, Hongqi Teng1, Yanming Wang1, Weijuan Pan4,FeiYu1, Lujian Liao4,LiLi5, Xin Ge6 and Ping Wang1 Mechanistic target of rapamycin mTOR complex 1 (mTORC1) plays a key role in the integration of various environmental signals to regulate cell growth and metabolism. mTORC1 is recruited to the lysosome where it is activated by its interaction with GTP-bound Rheb GTPase. However, the regulatory mechanism of Rheb activity remains largely unknown. Here, we show that ubiquitination governs the nucleotide-bound status of Rheb. Lysosome-anchored E3 ligase RNF152 catalyzes Rheb ubiquitination and promotes its binding to the TSC complex. EGF enhances the deubiquitination of Rheb through AKT-dependent USP4 phosphorylation, leading to the release of Rheb from the TSC complex. Functionally, ubiquitination of Rheb is linked to mTORC1-mediated signaling and consequently regulates tumor growth. Thus, we propose a mechanistic model whereby Rheb–mediated mTORC1 activation is dictated by a dynamic opposing act between Rheb ubiquitination and deubiquitination that are catalyzed by RNF152 and USP4 respectively. Cell Research (2019) 29:136–150; https://doi.org/10.1038/s41422-018-0120-9 INTRODUCTION GATOR1 is identified as a GAP (GTPase-activating protein) for The mechanistic target of rapamycin (mTOR) is a conserved Rag10. Senstrin2 mediates mTORC1 activity by acting as a GDI serine/threonine protein kinase in all eukaryotes that incorporates (guanine nucleotide dissociation inhibitor) for Rag11 or a protein various intracellular and extracellular signals including growth partener with GATOR212,13. -

Perkinelmer Genomics to Request the Saliva Swab Collection Kit for Patients That Cannot Provide a Blood Sample As Whole Blood Is the Preferred Sample
Autism and Intellectual Disability TRIO Panel Test Code TR002 Test Summary This test analyzes 2429 genes that have been associated with Autism and Intellectual Disability and/or disorders associated with Autism and Intellectual Disability with the analysis being performed as a TRIO Turn-Around-Time (TAT)* 3 - 5 weeks Acceptable Sample Types Whole Blood (EDTA) (Preferred sample type) DNA, Isolated Dried Blood Spots Saliva Acceptable Billing Types Self (patient) Payment Institutional Billing Commercial Insurance Indications for Testing Comprehensive test for patients with intellectual disability or global developmental delays (Moeschler et al 2014 PMID: 25157020). Comprehensive test for individuals with multiple congenital anomalies (Miller et al. 2010 PMID 20466091). Patients with autism/autism spectrum disorders (ASDs). Suspected autosomal recessive condition due to close familial relations Previously negative karyotyping and/or chromosomal microarray results. Test Description This panel analyzes 2429 genes that have been associated with Autism and ID and/or disorders associated with Autism and ID. Both sequencing and deletion/duplication (CNV) analysis will be performed on the coding regions of all genes included (unless otherwise marked). All analysis is performed utilizing Next Generation Sequencing (NGS) technology. CNV analysis is designed to detect the majority of deletions and duplications of three exons or greater in size. Smaller CNV events may also be detected and reported, but additional follow-up testing is recommended if a smaller CNV is suspected. All variants are classified according to ACMG guidelines. Condition Description Autism Spectrum Disorder (ASD) refers to a group of developmental disabilities that are typically associated with challenges of varying severity in the areas of social interaction, communication, and repetitive/restricted behaviors.