Zs2sess5zsis Skeletal, Digestive, Or Urogenital Systems, It Becomes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

3 Embryology and Development
BIOL 6505 − INTRODUCTION TO FETAL MEDICINE 3. EMBRYOLOGY AND DEVELOPMENT Arlet G. Kurkchubasche, M.D. INTRODUCTION Embryology – the field of study that pertains to the developing organism/human Basic embryology –usually taught in the chronologic sequence of events. These events are the basis for understanding the congenital anomalies that we encounter in the fetus, and help explain the relationships to other organ system concerns. Below is a synopsis of some of the critical steps in embryogenesis from the anatomic rather than molecular basis. These concepts will be more intuitive and evident in conjunction with diagrams and animated sequences. This text is a synopsis of material provided in Langman’s Medical Embryology, 9th ed. First week – ovulation to fertilization to implantation Fertilization restores 1) the diploid number of chromosomes, 2) determines the chromosomal sex and 3) initiates cleavage. Cleavage of the fertilized ovum results in mitotic divisions generating blastomeres that form a 16-cell morula. The dense morula develops a central cavity and now forms the blastocyst, which restructures into 2 components. The inner cell mass forms the embryoblast and outer cell mass the trophoblast. Consequences for fetal management: Variances in cleavage, i.e. splitting of the zygote at various stages/locations - leads to monozygotic twinning with various relationships of the fetal membranes. Cleavage at later weeks will lead to conjoined twinning. Second week: the week of twos – marked by bilaminar germ disc formation. Commences with blastocyst partially embedded in endometrial stroma Trophoblast forms – 1) cytotrophoblast – mitotic cells that coalesce to form 2) syncytiotrophoblast – erodes into maternal tissues, forms lacunae which are critical to development of the uteroplacental circulation. -

Patent Ductus Arteriosus About This Factsheet the Normal Heart
Understanding your child’s heart Patent ductus arteriosus About this factsheet The normal heart This factsheet is for parents of babies and children who The heart is a muscular pump which pumps blood through the have patent ductus arteriosus (PDA), which is also known as body and lungs. There are four chambers in the heart. The two persistent arterial duct. upper ones are called the right atrium and left atrium. These are separated by a wall called the atrial septum. The two lower It explains: chambers are called the right and left ventricles, and are separated • what patent ductus arteriosus is and how it is diagnosed by a wall called the ventricular septum. • how patent ductus arteriosus is treated • the benefits and risks of treatments. On each side of the heart, blood passes from the atrium, through a heart valve – the tricuspid valve on the right, and the mitral valve This factsheet does not replace the advice that doctors or on the left – into the ventricle. The ventricles are the main pumping nurses may give you, but it should help you to understand chambers of the heart. Each ventricle pumps blood out into an artery. what they tell you. The right ventricle pumps blood – blue in the illustration – into the pulmonary artery (the blood vessel that takes blood to the lungs). The left ventricle pumps blood – red in the illustration – into the aorta (the blood vessel that takes blood to the rest of the body). Blood flows from the right side of the heart, through the pulmonary valve into the pulmonary artery, and then to the lungs where it picks up oxygen. -
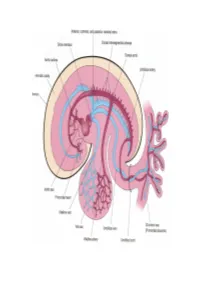
Development of HEART 4-VEINS
Development of brachiocephalic veins 1. Right brachiocephalic vein is formed by cranial part of right anterior cardinal vein and 2. Left brachiocephalic is formed by cranial part of left anterior cardinal vein and the interant.cardinal anastomosis. Development of superior vena cava 1. The part up to the opening of vena azygos develops from caudal part of right ant.cardinal vein and 2. The part below the opening (intrapericardial part) is formed by the right common cardinal vein. Development of azygos and hemiazygos veins A. 1. Vena azygos develops from right azygos line vein and 2. The arch of vena azygos is formed by the cranial end of right postcardinal vein. B. Hemiazygos veins are formed by the left azygos line vein. Development of Inferior vena cava Inferior vena cava is formed, from below upwards by: 1. Begins by the union of the two common iliac veins (postcardinal veins), 2. Right supracardinal, 3. Right supra-subcardinal anastomosis, 4. Right subcardinal, 5. New formation (hepatic segment) and 6. Hepatocardiac channel (terminal part of right vitelline vein). Congenital anomalies • Double inferior vena cava • Absence • Left SVC • Double SVC DEVELOPMENT OF PORTAL VEIN 1. The portal vein is formed behind the neck of pancreas by the union of superior mesentric and splenic vein to the left vitelline vein. 2. The part of the portal vein which is behind the Ist part of duodenum is formed by middle dorsal transverse anastomosis. 3. Part of portal vein which is in the free margin of lesser omentum is formed by cranial or distal part of right vitelline vein. -

Fetal Circulation
The Fetal Circulation Dr. S. Mathieu, Specialist Registrar in Anaesthesia Dr. D. J. Dalgleish, Consultant Anaesthetist Royal Bournemouth and Christchurch Hospitals Trust, UK Questions 1. In the fetal circulation: a) There are two umbilical arteries and one umbilical vein? b) Over 90% of blood passes the liver via the ductus venosus c) The foramen ovale divides the left and right ventricle d) The umbilical artery carries oxygenated blood from the placenta to the fetus e) The foramen ovale allows oxygenated blood to bypass the pulmonary circulation 2. In the fetal circulation: a) The oxygen dissociation curve of fetal haemoglobin is shifted to the left compared with adult haemoglobin ensuring oxygen delivery to the fetus despite low oxygen partial pressures b) It is the presence of the ductus arteriosus and large pulmonary vascular resistance which ensures most of the right ventricular output passes into the aorta c) The patency of the ductus arteriosus is maintained by high oxygen tensions d) The patency of the ductus arteriosus is maintained by the vasodilating effects of prostaglandin G2 e) 2,3-DPG levels are higher in fetal haemoglobin compared with adult haemaglobin 3. Changes at birth include: a) a fall in pulmonary vascular resistance b) a rise in systemic vascular resistance with clamping of the cord c) an increase in hypoxic pulmonary vasoconstriction d) a rise in left atrial pressure e) closure of the ductus arteriosus within 24 hours 4. The following congenital heart lesions are cyanotic: a) Ventricular septal defect b) Atrial septal defect c) Patent ductus arteriosus d) Tetralogy of Fallot e) Transposition of the great arteries MCQ answers at end Key points • The fetal circulation supplies the fetal tissues with oxygen and nutrients from the placenta. -

The Pattern and Mechanisms of Response to Oxygen by the Ductus Arteriosus and Umbilical Artery
Pediat. Res. 6: 693-700 (1972) Acetylcholine neonate atropine prematurity bradykinin sympathetic nervous system ductus arteriosus The Pattern and Mechanisms of Response to Oxygen by the Ductus Arteriosus and Umbilical Artery INGRID OBERHANSLI-WEISS, MICHAEL A. HEYMANN1391, ABRAHAM M. RUDOLPH, AND KENNETH L. MELMON Cardiovascular Research Institute and Departments of Pediatrics, Physiology and Pharmacology, University of California San Francisco, San Francisco, California, USA Extract Response of the ductus arteriosus and umbilical artery to changes in oxygen tension, to acetylcholine, and to sympathetic and parasympathetic blocking agents was studied in vitro in isolated rings obtained from 22 fetal lambs of 98- to 147-day gestation. After stabilization of tension at a baseline level (0.3-0.7 g) in a PO2 environment of 35-45 mm Hg, both increase of the PO2 to 550 mm Hg and decrease of the PO2 to 8 mm Hg of the bathing solution produced constriction. The mean maximal tension developed by the ductus arteriosus.was 3.91 g at high PO2 and 3.87 g at low POr The increase in maximal tension developed with advancing gestation was also similar at both high and low POj. At P02 levels of 8-550 mm Hg, acetylcholine produced a further increase in tension, whereas bradykinin only produced an increase in tension at high PO2- Alpha and beta sympathetic blockade had no effect on the constrictor response to oxygen. Atropine relaxed the ductus arteriosus and umbilical artery at both high and low Po2 levels; the degree of relaxation was related to drug concentration. Acetylcholin- esterase also relaxed the ductus arteriosus constricted by oxygen. -

Echocardiographic Follow-Up of Patent Foramen Ovale and the Factors Affecting Spontaneous Closure
Acta Cardiol Sin 2016;32:731-737 Brief Report doi: 10.6515/ACS20160205A Echocardiographic Follow-Up of Patent Foramen Ovale and the Factors Affecting Spontaneous Closure Ali Yildirim,1 Alperen Aydin,2 Tevfik Demir,1 Fatma Aydin,2 Birsen Ucar1 and Zubeyir Kilic1 Background: The aim of the present study was to evaluate the echocardiographic follow-up of patent foramen ovale, which is considered a potential etiological factor in various diseases, and to determine the factors affecting spontaneous closure. Methods: Between January 2000 and June 2012, records of 918 patients with patent foramen ovale were retrospectively reviewed. Patency of less than 3 mm around the fossa ovalis is called patent foramen ovale. Patients with cyanotic congenital heart diseases, severe heart valve disorders and severe hemodynamic left to right shunts were excluded from the study. The patients were divided into three groups based on age; 1 day-1 monthingroup1,1month-12monthsingroup2,andmorethan12monthsingroup3. Results: Of the 918 patients, 564 (61.4%) had spontaneous closure, 328 (35.8%) had patent foramen ovale continued, 15 (1.6%) patients had patent foramen ovale enlarged to 3-5 mm, 6 patients were enlarged to 5-8 mm, and in one patient patent foramen ovale reached to more than 8 mm size. Defect was spontaneously closed in 65.9% of the patients in group 1, 66.7% of the patients in group 2, and 52.3% of the patients in group 3. There was a negative correlation between the age of diagnosis and spontaneous closure (p < 0.05). Gender, prematurity and coexisting malformations such as patent ductus arteriosus and atrial septal aneurysm did not have any effect on spontaneous closure of patent foramen ovale (p > 0.05). -

Has a Role in Cardiovascular and Placental Development and Is a Binding Partner of the Α4 Integrin
Abl-interactor-1 (Abi1) has a role in cardiovascular and placental development and is a binding partner of the α4 integrin Colleen Ringa,1, Mark H. Ginsbergb, Jacob Halingb, and Ann Marie Pendergasta,1 aDepartment of Pharmacology and Cancer Biology, Duke University Medical Center, Durham, NC 27710; and bDepartment of Medicine, University of California at San Diego, La Jolla, CA 92093 Edited* by Stephen P. Goff, Columbia University College of Physicians and Surgeons, New York, NY, and approved November 30, 2010 (received for review August 19, 2010) Dynamic signals linking the actin cytoskeleton and cell adhesion properties among integrin subunits in that α4 predominantly receptors are essential for morphogenesis during development accumulates at the leading edge of migrating cells, rather than at and normal tissue homeostasis. Abi1 is a central regulator of actin focal adhesions. Moreover, α4 expression is associated with polymerization through interactions with multiple protein com- protrusive activity and enhanced cell migration; however, the plexes. However, the in vivo role of Abi1 remains to be defined. pathways that link α4 integrin to the actin-regulatory machinery The α4 integrin adhesion receptor is associated with enhanced at the leading edge have remained elusive. Here we identify Abi1 protrusive activity and regulation of directional cell migration. as a target of α4 integrin that positively regulates membrane Among integrin subunits, α4 exhibits unique properties in that it protrusion by promoting actin polymerization at sites of integrin predominantly accumulates at the leading edge of migrating cells; engagement. however, the pathways that link the actin-regulatory machinery to The Abi family proteins, Abi1 and Abi2, were originally id- α4 at the leading edge have remained elusive. -

6 Development of the Great Vessels and Conduction Tissue
Development of the Great Vessels and Conduc6on Tissue Development of the heart fields • h:p://php.med.unsw.edu.au/embryology/ index.php?6tle=Advanced_-_Heart_Fields ! 2 Septa6on of the Bulbus Cordis Bulbus Cordis AV Canal Ventricle Looking at a sagital sec6on of the heart early in development the bulbus cordis is con6nuous with the ventricle which is con6nuous with the atria. As the AV canal shiOs to the right the bulbus move to the right as well. Septa6on of the Bulbus Cordis A P A P The next three slides make the point via cross sec6ons that the aorta and pulmonary arteries rotate around each other. This means the septum between them changes posi6on from superior to inferior as well. Septa6on of the Bulbus Cordis P A A P Septa6on of the Bulbus Cordis P A P A Migra6on of neural crest cells Neural crest cells migrate from the 3ed, 4th and 6th pharyngeal arches to form some of the popula6on of cells forming the aor6copulmonary septum. Septa6on of the Bulbus Cordis Truncal (Conal) Swellings Bulbus Cordis The cardiac jelly in the region of the truncus and conus adds the neural crest cells and expands as truncal swellings. Septa6on of the Bulbus Cordis Aorticopulmonary septum These swellings grow toward each other to meet and form the septum between the aorta and pulmonary artery. Aorta Pulmonary Artery Septa6on of the Bulbus Cordis Anterior 1 2 3 1 2 3 The aor6copulmonary septum then rotates as it moves inferiorly. However, the exact mechanism for that rota6on remains unclear. Septa6on of the Bulbus Cordis Aorta Pulmonary Artery Conal Ridges IV Foramen Membranous Muscular IV Endocarial Septum Interventricular Cushion Septum However, the aor6copulmonary septum must form properly for the IV septum to be completed. -

Equine Placenta – Marvelous Organ and a Lethal Weapon
Equine placenta – marvelous organ and a lethal weapon Malgorzata Pozor, DVM, PhD, Diplomate ACT Introduction Placenta has been defined as: „an apposition between parent (usually maternal) and fetal tissue in order to establish physiological exchange” (1). Another definition of this important organ was proposed by Steven and Morris: „a device consisting of one or more transport epithelia located between fetal and maternal blood supply” (2). The main function of placenta is to provide an interface between the dam and the the fetus and to allow the metabolic exchange of the the nutrients, oxygen and waste material. The maternal circulation is brought into a close apposition to the fetal circulation, while a separation of these two circulatory systems remain separated (3). A degree and complexity of this „intimate relationship” varies greately between species mostly due to the structural diversity of the extraembryonic membranes of the vertebrates. The early feto-maternal exchange in the equine pregnancy is established as early as on day 22 after fertilization. The fetal and choriovitellin circulations are already present, the capsule ruptures and the allantois is already visible (4). The allantois starts expanding by day 32 and vascularizes approximately 90% of the chorion and fuses with it to form chorioallantois by day 38 of gestation (5). The equine placenta continues increasing its complexity till approximately day 150 of gestation. Equids have epitheliochorial placenta, there are six leyers separating maternal and fetal circulation, and there are no erosion of the luminal, maternal epithelium, like in ruminants (6). Thousands of small chorionic microvilli develop and penetrate into endometrial invaginations. -

COMMENTARY the First Evidence of the Tumor-Induced Angiogenesis in Vivo by Using the Chorioallantoic Membrane Assay Dated 1913
Leukemia (2004) 18, 1350–1351 & 2004 Nature Publishing Group All rights reserved 0887-6924/04 $30.00 www.nature.com/leu COMMENTARY The first evidence of the tumor-induced angiogenesis in vivo by using the chorioallantoic membrane assay dated 1913 Domenico Ribatti1 1Department of Human Anatomy and Histology, University of Bari Medical School, Bari, Italy Leukemia (2004) 18, 1350–1351. doi:10.1038/sj.leu.2403411 tional characterization of the immune system in the chick Published online 17 June 2004 embryo. Early lymphoid cells deriving from the yolk sac and spleen are usually recognizable in the thymus on day 8 and in Virchow, the founder of pathological anatomy, drew attention to the bursa of Fabricius on day 11.6 Thymus cells are present by the huge number of blood vessels in a tumor mass as long ago as day 11 and cell-mediated immunity has been demonstrated by 1865. Tumor vascularization was first studied systematically by day 13–14.7 The chick embryo and the nude mouse are 1 Goldman, who described the vasoproliferative response of the immunological incompetent hosts and do not reject tissues organ in which a tumor develops as follows: ‘The normal blood from a foreign source. Indeed, the chick embryo cannot mount vessels of the organs in which the tumor is developing are an ‘immune’ response to foreign tumor cells until well after day disturbed by chaotic growth, there is a dilatation and spiralling 12, but it can respond to tumor cells by infiltration of monocytes of the affected vessels, marked capillary budding and new vessel and inflammatory-like cells such as avian heterophils. -

From Trophoblast to Human Placenta
From Trophoblast to Human Placenta (from The Encyclopedia of Reproduction) Harvey J. Kliman, M.D., Ph.D. Yale University School of Medicine I. Introduction II. Formation of the placenta III. Structure and function of the placenta IV. Complications of pregnancy related to trophoblasts and the placenta Glossary amnion the inner layer of the external membranes in direct contact with the amnionic fluid. chorion the outer layer of the external membranes composed of trophoblasts and extracellular matrix in direct contact with the uterus. chorionic plate the connective tissue that separates the amnionic fluid from the maternal blood on the fetal surface of the placenta. chorionic villous the final ramification of the fetal circulation within the placenta. cytotrophoblast a mononuclear cell which is the precursor cell of all other trophoblasts. decidua the transformed endometrium of pregnancy intervillous space the space in between the chorionic villi where the maternal blood circulates within the placenta invasive trophoblast the population of trophoblasts that leave the placenta, infiltrates the endo– and myometrium and penetrates the maternal spiral arteries, transforming them into low capacitance blood channels. Sunday, October 29, 2006 Page 1 of 19 From Trophoblasts to Human Placenta Harvey Kliman junctional trophoblast the specialized trophoblast that keep the placenta and external membranes attached to the uterus. spiral arteries the maternal arteries that travel through the myo– and endometrium which deliver blood to the placenta. syncytiotrophoblast the multinucleated trophoblast that forms the outer layer of the chorionic villi responsible for nutrient exchange and hormone production. I. Introduction The precursor cells of the human placenta—the trophoblasts—first appear four days after fertilization as the outer layer of cells of the blastocyst. -

Utilisation of Chick Embryo Chorioallantoic Membrane As a Model Platform for Imaging-Navigated Biomedical Research
cells Review Utilisation of Chick Embryo Chorioallantoic Membrane as a Model Platform for Imaging-Navigated Biomedical Research Lei Chen 1 , Shuncong Wang 1 , Yuanbo Feng 1, Jinyong Zhang 2,3, Yuqing Du 2, Jiang Zhang 4, Chantal Van Ongeval 1, Yicheng Ni 1,* and Yue Li 2,* 1 KU Leuven, Biomedical Group, Campus Gasthuisberg, 3000 Leuven, Belgium; [email protected] (L.C.); [email protected] (S.W.); [email protected] (Y.F.); [email protected] (C.V.O.) 2 Shanghai Key Laboratory of Molecular Imaging, Shanghai University of Medicine and Health Sciences, Shanghai 201318, China; [email protected] (J.Z.); [email protected] (Y.D.) 3 School of Medical Instrument and Food Engineering, University of Shanghai for Science & Technology, Shanghai 200093, China 4 Faculty of Agricultural Biotechnology and Ecotechnology, Shanghai Vocational College of Agriculture and Forestry, Shanghai 201600, China; [email protected] * Correspondence: [email protected] (Y.N.); [email protected] (Y.L.) Abstract: The fertilised chick egg and particularly its chorioallantoic membrane (CAM) have drawn continuing interest in biomedicine and bioengineering fields, especially for research on vascular study, cancer, drug screening and development, cell factors, stem cells, etc. This literature review sys- temically introduces the CAM’s structural evolution, functions, vascular features and the circulation system, and cell regulatory factors. It also presents the major and updated applications of the CAM in assays for pharmacokinetics and biodistribution, drug efficacy and toxicology testing/screening in preclinical pharmacological research. The time course of CAM applications for different assays Citation: Chen, L.; Wang, S.; Feng, Y.; and their advantages and limitations are summarised.