Synthesis and Reactions of Substituted Cyclopropanols Kenneth Lee Eilers Iowa State University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evaluated Gas Phase Basicities and Proton Affinities of Molecules; Heats of Formation of Protonated Molecules
Evaluated Gas Phase Basicities and Proton Affinities of Molecules; Heats of Formation of Protonated Molecules Cite as: Journal of Physical and Chemical Reference Data 13, 695 (1984); https://doi.org/10.1063/1.555719 Published Online: 15 October 2009 Sharon G. Lias, Joel F. Liebman, and Rhoda D. Levin ARTICLES YOU MAY BE INTERESTED IN Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update Journal of Physical and Chemical Reference Data 27, 413 (1998); https:// doi.org/10.1063/1.556018 Density-functional thermochemistry. III. The role of exact exchange The Journal of Chemical Physics 98, 5648 (1993); https://doi.org/10.1063/1.464913 A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu The Journal of Chemical Physics 132, 154104 (2010); https://doi.org/10.1063/1.3382344 Journal of Physical and Chemical Reference Data 13, 695 (1984); https://doi.org/10.1063/1.555719 13, 695 © 1984 American Institute of Physics for the National Institute of Standards and Technology. Evaluated Gas Phase Basicities and Proton Affinities of Molecules; Heats of Formation of Protonated Molecules Sharon G. Lias Center for Chemical Physics, National Bureau of Standards, Gaithersburg, MD 20899 Joel F. Liebman Department of Chemistry, University of Maryland Baltimore County, Catonsville, MD 21228 and Rhoda D. Levin Center for Chemical Physics. National Bureau of Standards, Gaithersburg, MD 20899 The available data on gas phase basicities and proton affinities of molecules are compiled and evaluated. Tables giving the molecules ordered (1) according to proton affinity and (2) according to empirical formula, sorted alphabetically are provided. -

WO 2016/022464 Al 11 February 2016 (11.02.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/022464 Al 11 February 2016 (11.02.2016) P O P C T (51) International Patent Classification GELMAN, Leonid; 260 E. Grand Avenue, 2nd Floor, A61K 31/7068 (2006.01) A61K 31/519 (2006.01) South San Francisco, CA 94080 (US). SMITH, David, A61K 31/4184 (2006.01) A61K 31/166 (2006.01) Bernard; 260 E. Grand Avenue, 2nd Floor, South San A61K 31/675 (2006.01) A61K 31/41 (2006.01) Francisco, CA 94080 (US). WANG, Guangyi; 260 E. A61K 31/513 (2006.01) A61K 38/21 (2006.01) Grand Avenue, 2nd Floor, South San Francisco, CA 94080 A61K 31/506 (2006.01) A61K 38/16 (2006.01) (US). A61K 31/437 (2006.01) A61K 39/155 (2006.01) (74) Agent: MILLER, Kimberly, J.; Knobbe Martens Olson A61K 31/4709 (2006.01) A61K 39/42 (2006.01) & Bear, LLP, 2040 Main Street, 14th Floor, Irvine, CA A61K 31/4188 (2006.01) A61P 31/14 (2006.01) 92614 (US). A61K 31/517 (2006.01) A61P 11/00 (2006.01) (81) Designated States (unless otherwise indicated, for every (21) International Application Number: PCT/US20 15/043402 kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (22) International Filing Date: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, 3 August 2015 (03.08.2015) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, English (25) Filing Language: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (26) Publication Language: English MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (30) Priority Data: SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 62/033,55 1 5 August 2014 (05.08.2014) US TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Revised Group Additivity Values for Enthalpies of Formation (At 298 K) of Carbon– Hydrogen and Carbon–Hydrogen–Oxygen Compounds
Revised Group Additivity Values for Enthalpies of Formation (at 298 K) of Carbon– Hydrogen and Carbon–Hydrogen–Oxygen Compounds Cite as: Journal of Physical and Chemical Reference Data 25, 1411 (1996); https://doi.org/10.1063/1.555988 Submitted: 17 January 1996 . Published Online: 15 October 2009 N. Cohen ARTICLES YOU MAY BE INTERESTED IN Additivity Rules for the Estimation of Molecular Properties. Thermodynamic Properties The Journal of Chemical Physics 29, 546 (1958); https://doi.org/10.1063/1.1744539 Critical Evaluation of Thermochemical Properties of C1–C4 Species: Updated Group- Contributions to Estimate Thermochemical Properties Journal of Physical and Chemical Reference Data 44, 013101 (2015); https:// doi.org/10.1063/1.4902535 Estimation of the Thermodynamic Properties of Hydrocarbons at 298.15 K Journal of Physical and Chemical Reference Data 17, 1637 (1988); https:// doi.org/10.1063/1.555814 Journal of Physical and Chemical Reference Data 25, 1411 (1996); https://doi.org/10.1063/1.555988 25, 1411 © 1996 American Institute of Physics for the National Institute of Standards and Technology. Revised Group Additivity Values for Enthalpies of Formation (at 298 K) of Carbon-Hydrogen and Carbon-Hydrogen-Oxygen Compounds N. Cohen Thermochemical Kinetics Research, 6507 SE 31st Avenue, Portland, Oregon 97202-8627 Received January 17, 1996; revised manuscript received September 4, 1996 A program has been undertaken for the evaluation and revision of group additivity values (GAVs) necessary for predicting, by means of Benson's group additivity method, thermochemical properties of organic molecules. This review reports on the portion of that program dealing with GAVs for enthalpies of formation at 298.15 K (hereinafter abbreviated as 298 K) for carbon-hydrogen and carbon-hydrogen-oxygen compounds. -
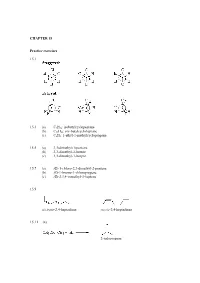
CHAPTER 15 Practice Exercises 15.1 15.3 (A) C9H18
CHAPTER 15 Practice exercises 15.1 15.3 (a) C9H18: isobutylcyclopentane (b) C11H22: sec-butylcycloheptane (c) C6H2: 1-ethyl-1-methylcyclopropane 15.5 (a) 3,3-dimethyl-1-pentene (b) 2,3-dimethyl-2-butene (c) 3,3-dimethyl-1-butyne 15.7 (a) (E)-1-chloro-2,3-dimethyl-2-pentene (b) (Z)-1-bromo-1-chloropropene (c) (E)-2,3,4-trimethyl-3-heptene 15.9 cis,trans-2,4-heptadiene cis,cis-2,4-heptadiene 15.11 (a) 2-iodopropane (b) 1-iodo-1-methylcyclohexane 15.13 CH3 CH3 + HI I Step 1: Protonation of the alkene to give the most stable 3˚ carbocation intermediate: slow step + CH3 HI CH3 I rate-determining step Step 2: Nucleophilic attack of the iodide anion on the 3˚ carbocation intermediate to give the product: CH + 3 CH3 I I 15.15 H2O CH3 CH3 + H3O OH Step 1: Protonation of the alkene to give the most stable 3˚ carbocation intermediate: slow step H + + CH3 HOH CH3 O H rate-determining H step Step 2: Nucleophilic attack of the water on the 3˚ carbocation intermediate to give the protonated alcohol: H H O+ H + CH3 O H CH3 Step 3: The protonated alcohol loses a proton to form the product: H + O H H CH3 + O + H3O CH3 H OH 15.17 (a) 2-phenyl-2-propanol (b) (E)-3,4-diphenyl-3-hexene (c) 3-methylbenzoic acid or m-methylbenzoic acid Review questions 15.1 A hydrocarbon is a compound composed only of hydrogen and carbon atoms. 15.3 In saturated hydrocarbons, each carbon is bonded to four other atoms, either hydrogen or carbon atoms. -

Maximum Topological Distances Based Indices As Molecular Descriptors for QSPR
Int. J. Mol. Sci. 2001, 2, 121-132 International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.org/ijms/ Maximum Topological Distances Based Indices as Molecular Descriptors for QSPR. 4. Modeling the Enthalpy of Formation of Hydrocarbons from Elements Andrés Mercader 1, Eduardo A. Castro 1,* and Andrey A. Toropov2 1 CEQUINOR, Departamento de Química, Facultad de Ciencias Exactas, Universidad Nacional de la Plata, C.C. 962, La Plata 1900, Argentina. e-mail: [email protected] 2 Andrey A. Toropov, Vostok Innovation Company, S. Azimstreet 4, 100047 Tashkent, Uzbekistan. e-mail: [email protected] * Author to whom correspondence should be addressed. e-mail: [email protected] Received: 9 April 2001 Accepted: 15 June 2001/ Published: 28 June 2001 Abstract: The enthalpy of formation of a set of 60 hydroarbons is calculated on the basis of topological descriptors defined from the distance and detour matrices within the realm of the QSAR/QSPR theory. Linear and non-linear polynomials fittings are made and results show the need to resort to higher-order regression equations in order to get better concordances between theoretical results and experimental available data. Besides, topological indices computed from maximum order distances seems to yield rather satisfactory predictions of heats of formation for hydrocarbons. Keywords: QSPR theory, Detour matrix, Enthalpy of formation, Hydrocarbons, Topologi- cal indices 1. Introduction Graphs have found considerable employment in several chemistry fields, particularly in modeling molecular structure [1-10]. The applications of graphs to the study of structure-property relationships implies the representation of molecules by selected molecular descriptors, often referred to as topo- logical indices [11,12]. -

Trend Analysis for Atmospheric Hydrocarbon Partitioning Using Continuous Thermodynamics
AUGUST 2005 HARSTAD 2977 Trend Analysis for Atmospheric Hydrocarbon Partitioning Using Continuous Thermodynamics K. HARSTAD Jet Propulsion Laboratory, California Institute of Technology, Pasadena, California (Manuscript received 22 April 2004, in final form 31 January 2005) ABSTRACT The partitioning of atmospheric hydrocarbons into vapor and condensed phases when the species count is large is considered using the formalism of continuous thermodynamics. The vapor saturation pressures and condensate species distribution are parameterized using the species normal boiling temperatures. Qualitative trends in activity coefficient values and phase equilibrium behavior that are relevant to the outer planets and Titan are discussed in terms of a much simplified perspective on these aspects of partitioning. The trends found are generally consistent with those from other published atmospheric model results. 1. Introduction with particular emphasis on extraterrestrial bodies Hydrocarbons (and possibly derivative, i.e., oxidized without oxidizing environments. These models are in species) are commonly present in small amounts in general, very complex, involving partly the kinetics of a planetary atmospheres (e.g., Earth, the outer planets, large number of chemical reactions as well as turbulent Titan), and may attain a relatively wide range of carbon diffusion of chemical species in the atmosphere (Do- number. Abiotic generation is possible by chemical in- brijevic et al. 2003; Lebonnois et al. 2001; Lee et al. 2000; Moses et al. 2000; Raulin and Bruston 1996). De- teraction of CO (or CO2) with H2 (Raulin and Bruston 1996; Zolotov and Shock 1999) and, more effectively, spite the relative complexity of the models in the cited references, a totally complete and precise description of photochemistry of CH4 (Dobrijevic et al. -

Cometabolic Degradation of Polycyclic Aromatic Hydrocarbons (Pahs) and Aromatic Ethers by Phenol- and Ammonia-Oxidizing Bacteria
AN ABSTRACT OF THE DISSERTATION OF Soon Woong Chang for the degree of Doctor of Philosophy in Civil Engineering presented on November 11, 1997. Title: Cometabolic Degradation of Polycyclic Aromatic Hydrocarbons (PAHs) and Aromatic Ethers by Phenol- and Ammonia-Oxidizing Bacteria Redacted for Privacy Abstract approved: /Kenneth J. Williamson Cometabolic biodegradation processes are potentially useful for the bioremediation of hazardous waste sites. In this study the potential application of phenol- oxidizing and nitrifying bacteria as "priming biocatalysts" was examined in the degradation of polycyclic aromatic hydrocarbons (PAHs), aryl ethers, and aromatic ethers. We observed that a phenol-oxidizing Pseudomonas strain cometabolically degrades a range of 2- and 3-ringed PAHs. A sequencing batch reactor (SBR) was used to overcome the competitive effects between two substrates and the SBR was evaluated as a alternative technology to treat mixed contaminants including phenol and PAHs. We also have demonstrated that the nitrifying bacterium Nitrosomonas europaea can cometabolically degrade a wide range polycyclic aromatic hydrocarbons (PAHs), aryl ethers and aromatic ethers including naphthalene, acenaphthene, diphenyl ether, dibenzofuran, dibenzo-p-dioxin, and anisole. Our results indicated that all the compounds are transformed by N. europaea and that several unusual reactions are involved in these reactions. In the case of naphthalene oxidation, N. europaea generated predominantly 2 naphthol whereas other monooxygenases generate 1-naphthol as the major product. In the case of dibenzofuran oxidation, 3-hydroxydibenzofuran initially accumulated in the reaction medium and was then further transformed to 3-hydroxy nitrodibenzofuran in a pH- and nitrite-dependent abiotic reaction. A similar abiotic transformation reaction also was observed with other hydroxylated aryl ethers and PAHs. -

27-2-Erraigkarl HASS at TORNEYS
Dec. 24, 1968 A. LENZ ETAL 3,418,383 PRODUCTION OF ALKALI METAL, ALCOHOLATES Filed Nov. 8, 1966 g-Lower and higher olcohol vapors Lower Olcohol Condenser X Distillation column Reactor column Receiver Zy -IO 3 lower alcohol Higher olcohol Olcoholote vopor higher olcohol Liquid alcohol Solution S mixture Resevoir Superheater 15 a - 13 Higher alcohol Pump1 D HigherOlcohol olcoholote NVENTORS ARNOLD LENZ OTTO BLEH 27-2-erraigKARL HASS AT TORNEYS. 3,418,383 United States Patent Office Patented Dec. 24, 1968 2 e.g., isopropanol, butanol - (2), pentanol - (2), pentanol 3,418,383 (3), 2-methylbutanol - (3) and the like; and the tertiary PRODUCTION OF ALKALIMETAL ALCOHOLATES alcohols, e.g., 2-methylpropanol-(2), 2-methylbutanol-(2) Arnold Lenz, Gerstenkamp, Otto Bleh, Bergheim, and and the like. In addition to the univalent aliphatic alco Karl Hass, Niederkassel, Germany, assignors to Dyna hols, cycloaliphatic alcohols can be interchanged such as: mit Nobel Aktiengesellschaft cyclopropanol, cyclobutanol, cyclopentanol, cyclohexanol, Claims Filedpriority, Nov. application 8, 1966, Germany,Ser. No. 594,333 Nov. 10, 1965, and the alkyl-substituted derivatives thereof having a total D 48,614 of up to 12 carbon atoms. 9 Claims. (CI. 260-632) Both the alkali alcoholates of methanol and those of O ethanol can be used as starting products, and the alkali metal of the alcoholate can be sodium, potassium and ABSTRACT OF THE DISCLOSURE lithium, and, of course, cesium and rubidium as well. The improved interchange reaction between a lower The process of the invention is to be explained by alcoholate of an alkali metal and a higher alcohol to pro means of the annexed drawing which is a flow diagram of duce a higher alcoholate of the alkali metal, wherein the 15 the process. -

United States Patent Office Patented Jan
3,164,611 United States Patent Office Patented Jan. 5, 1965 1. 2 reaction, care must be taken in applying this method 3,164,611 to the oxidation of heat sensitive compounds. OXDATION OF PRIMARY AND SECONDARY AL COHOLS TO THE CORRESPONDING CARBONY These organic base-chromium trioxide complexes are CSCMPOUNDS (USNG A TERTARY AMENE particularly useful oxidizing agents for effecting the oxida (CERORySSJR, TROXDE COMPLEX tion of alcohols having at least one hydrogen atom at Lewis H. Sarett, Friscetos, N.J., assigaor to Merck & Co., tached to the carbon atom bearing the hydroxyl sub Inc., Rahway, N.J., a corporatioia of New Jersey Stituent, i.e., primary and secondary alcohols, to the No Drawing. Fied any 26, 1956, Ser. No. 686,463 corresponding carbonyl compounds. Thus, primary al 13 Caias. (C. 260-349.9) cohols are oxidized to aldehydes, and secondary alcohols IO are converted to ketones. This invention relates to a novel process for the oxida This method of oxidizing alcohols to the corresponding tion of chemical compounds, and more particularly to carbonyl compounds is generally applicable to all pri an improved method for the oxidation of primary and mary and secondary alcohols. Examples of such al Secondary alcohols to the corresponding carbonyl com cohols that might be mentioned are aliphatic alcohols pounds. 5 such as alkanals, alkenols, alkinois, polyhydric alkanols, This application is a continuation-in-part application polyhydric alkenols and polyhydric alkinols; aralkyl al of my application Serial No. 263,016, filed December cohols; aralkenyl alcohols; aralkinyl alcohols; alicyclic 22, 1951, now abandoned, and my copending application alcohols such as cycloalkyl, cycloalkenyl, cycloalkinyl, Serial No. -

A Review on Green Liquid Fuels for the Transportation Sector: a Prospect of Microbial Solutions to Climate Change
Biofuel Research Journal 23 (2019) 995-1024 Journal homepage: www.biofueljournal.com Review Paper A review on green liquid fuels for the transportation sector: a prospect of microbial solutions to climate change Hamed Kazemi Shariat Panahi1,2, Mona Dehhaghi1,2, James E. Kinder 3, Thaddeus Chukwuemeka Ezeji3,* 1 Faculty of Medicine and Health Sciences, Macquarie University, NSW, Australia. 2Department of Microbial Biotechnology, School of Biology and Centre of Excellence in Phylogeny of Living Organisms, College of Science, University of Tehran, Tehran, Iran. 3Department of Animal Sciences, Ohio State Agricultural Research and Development Center (OARDC), The Ohio State University, Wooster, USA. HIGHLIGHTS GRAPHICAL ABSTRACT Microbial-based biofuel as a promising waste-to- energy technology has been scrutinized. Microbial production of bio-jet fuel is possible through DSHC, AtJ, and GtL. Future application of ammonia as bio-fuel requires special design of ICE. Cons and pros of microbial liquid fuels over gasoline have been outlined. Conversion of microbial liquid fuel into fuel derivatives has been discussed. ARTICLE INFO ABSTRACT Article history: Environmental deterioration, global climate change, and consequent increases in pollution-related health problems among Received 12 July 2019 populations have been attributed to growing consumption of fossil fuels in particular by the transportation sector. Hence, Received in revised form 18 August 2019 replacing these energy carriers, also known as major contributors of greenhouse gas emissions, with biofuels have been regarded Accepted 20 August 2019 as a solution to mitigate the above-mentioned challenges. On the other hand, efforts have been put into limiting the utilization Available online 1 September 2019 of edible feedstocks for biofuels production, i.e., first generation biofuels, by promoting higher generations of these eco-friendly alternatives. -

Alcohol Oxidase from Hansenula Polymorpha
Alcohol Oxidase from Hansenula polymorpha Catalog Number A0438 Storage Temperature –20 C CAS RN 9073-63-6 Molecular mass:6 670 kDa (octomer, gel filtration) EC 1.1.3.13 Synonym: Alcohol:oxygen oxidoreductase Alcohol oxidase is a homooctomeric flavoprotein with eight equal subunits of 83 kDa; each of which contains Product Description a flavin adenine dinucleotide (FAD) molecule.6 Alcohol oxidase catalyzes the oxidation of short-chain, primary, aliphatic alcohols to the respective aldehydes. Cofactor:4 FAD, one molecule/subunit 7 RCH2OH + O2 RCHO + H2O2 Isoelectric point: 6.1 The enzyme has the highest affinity for methanol with pH Range:8 6.7–9.8 the affinity decreasing with increasing chain length of the alkyl (R) group. pH Optimum:8 8.5 5 Alcohol oxidase plays a major role in the metabolism of Ki (mM): methanol resulting in the formation of formaldehyde and methanol 6,500 has been detected in several genera of yeasts, such as Candida, Pichia, and Hansenula, that utilize methanol Inhibitors:3,8 as a sole carbon and energy source.1,2 1,4-butynediol (irreversible) 4-hydroxy-2- butynal Primarily localized in the peroxisome, alcohol oxidase cyclopropanol has also been found in the cytoplasm. Monomers are cyclopropanone (suicide substrate) synthesized in the cytosol and assembled into octomers formaldehyde, H2O2 (5–10 mM) in the peroxisome. Octomerization is thought to be hydroxylamine, KBr, KCN chaparone mediated.3 Alcohol oxidase is of interest for sodium azide, urea the study of protein translocation into peroxisomes.4 This product is purified from Hansenula polymorpha 4,5 KM (mM): and is supplied as an orange vacuum-dried powder. -

Chapter 21 Organic Chemistry
CHAPTER 21 ORGANIC CHEMISTRY Hydrocarbons 1. A hydrocarbon is a compound composed of only carbon and hydrogen. A saturated hydro- carbon has only carbon-carbon single bonds in the molecule. An unsaturated hydrocarbon has one or more carbon-carbon multiple bonds but may also contain carbon-carbon single bonds. A normal hydrocarbon has one chain of consecutively bonded carbon atoms. A branched hydrocarbon has at least one carbon atom not bonded to the end carbon of a chain of consecutively bonded carbon atoms. Instead, at least one carbon atom forms a bond to an inner carbon atom in the chain of consecutively bonded carbon atoms. 2. To determine the number of hydrogens bonded to the carbons in cyclic alkanes (or any alkane where they may have been omitted), just remember that each carbon has four bonds. In cycloalkanes, only the C−C bonds are shown. It is assumed you know that the remaining bonds on each carbon are C−H bonds. The number of C−H bonds is that number required to give the carbon four total bonds. 3. In order to form, cyclopropane and cyclobutane are forced to form bond angles much smaller than the preferred 109.5° bond angles. Cyclopropane and cyclobutane easily react in order to obtain the preferred 109.5° bond angles. 4. Aromatic hydrocarbons are a special class of unsaturated hydrocarbons based on the benzene ring. Benzene has the formula C6H6. It is a planar molecule (all atoms are in the same plane). Each carbon in benzene is attached to three other atoms; it exhibits trigonal planar geometry with 120° bond angles.