Xerox University Microfilms
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
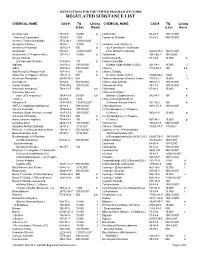
Regulated Substance List
INSTRUCTIONS FOR THE UNIFIED PROGRAM (UP) FORM REGULATED SUBSTANCE LIST CHEMICAL NAME CAS # TQ Listing CHEMICAL NAME CAS # TQ Listing (Lbs) Basis (Lbs) Basis Acetaldehyde 75-07-0 10,000 g Cantharidin 56-25-7 100/10,0001 * Acetone Cyanohydrin 75-86-5 1,000 Carbachol Chloride 51-83-2 500/10,0001 Acetone Thiosemicarbazide 1752-30-3 1,000/10,0001 Acetylene (Ethyne) 74-86-2 10,000 f Carbamic Acid, Methyl-,o- Acrolein (2-Propenal) 107-02-8 500 b (((2,4-Dimethyl-1,3-Dithiolan- Acrylamide 79-06-1 1,000/10,0001 2-YL) Methylene)Amino)- 26419-73-8 100/10,0001 Acrylonitrile (2- Propenenitrile) 107-13-1 10,000 b Carbofuran 1563-66-2 10/10,0001 Acrylyl Chloride Carbon Disulfide 75-15-0 10,000 b (2-Propenoyl Chloride) 814-68-6 100 b Carbon Oxysulfide Aldicarb 116-06-3 100/10,0001 (Carbon Oxide Sulfide (COS)) 463-58-1 10,000 f Aldrin 309-00-2 500/10,0001 Chlorine 7782-50-5 100 a,b Allyl Alcohol (2-Propen-1-ol) 107-18-6 1,000 b Chlorine Dioxide Allylamine (2-Propen-1-Amine) 107-11-9 500 b (Chlorine Oxide (ClO2)) 10049-04-4 1,000 c Aluminum Phosphide 20859-73-8 500 Chlorine Monoxide (Chlorine Oxide) 7791-21-1 10,000 f Aminopterin 54-62-6 500/10,0001 Chlormequat Chloride 999-81-5 100/10,0001 Amiton Oxalate 3734-97-2 100/10,0001 Chloroacetic Acid 79-11-8 100/10,0001 Ammonia, Anhydrous 2 7664-41-7 500 a,b Chloroform 67-66-3 10,000 b Ammonia, Aqueous Chloromethyl Ether (conc 20% or greater) 7664-41-7 20,000 a,b (Methane,Oxybis(chloro-) 542-88-1 100 b * Aniline 62-53-3 1,000 Chloromethyl Methyl Ether Antimycin A 1397-94-0 1,000/10,0001 (Chloromethoxymethane) -

Thermodynamic Hydricity of Small Borane Clusters and Polyhedral Closo-Boranes
molecules Article Thermodynamic Hydricity of Small Borane Clusters y and Polyhedral closo-Boranes Igor E. Golub 1,* , Oleg A. Filippov 1 , Vasilisa A. Kulikova 1,2, Natalia V. Belkova 1 , Lina M. Epstein 1 and Elena S. Shubina 1,* 1 A. N. Nesmeyanov Institute of Organoelement Compounds and Russian Academy of Sciences (INEOS RAS), 28 Vavilova St, 119991 Moscow, Russia; [email protected] (O.A.F.); [email protected] (V.A.K.); [email protected] (N.V.B.); [email protected] (L.M.E.) 2 Faculty of Chemistry, M.V. Lomonosov Moscow State University, 1/3 Leninskiye Gory, 119991 Moscow, Russia * Correspondence: [email protected] (I.E.G.); [email protected] (E.S.S.) Dedicated to Professor Bohumil Štibr (1940-2020), who unfortunately passed away before he could reach the y age of 80, in the recognition of his outstanding contributions to boron chemistry. Academic Editors: Igor B. Sivaev, Narayan S. Hosmane and Bohumír Gr˝uner Received: 6 June 2020; Accepted: 23 June 2020; Published: 25 June 2020 MeCN Abstract: Thermodynamic hydricity (HDA ) determined as Gibbs free energy (DG◦[H]−) of the H− detachment reaction in acetonitrile (MeCN) was assessed for 144 small borane clusters (up 2 to 5 boron atoms), polyhedral closo-boranes dianions [BnHn] −, and their lithium salts Li2[BnHn] (n = 5–17) by DFT method [M06/6-311++G(d,p)] taking into account non-specific solvent effect (SMD MeCN model). Thermodynamic hydricity values of diborane B2H6 (HDA = 82.1 kcal/mol) and its 2 MeCN dianion [B2H6] − (HDA = 40.9 kcal/mol for Li2[B2H6]) can be selected as border points for the range of borane clusters’ reactivity. -

Minutesbasel Approved
Approved minutes, Basel 2018 International Union of Pure and Applied Chemistry Division VIII Chemical Nomenclature and Structure Representation Approved minutes for Division Committee Meeting Basel, Switzerland, 13–14 August, 2018 1. Welcome, introductory remarks and housekeeping announcements Alan Hutton (ATH) welcomed everybody to the meeting, extending a special welcome to those who were attending the Division Committee (DC) meeting for the first time, and noting the presence of three former Presidents of the Division, as well as the current Secretary General of IUPAC. He described the house rules and arrangements for the course of the meeting. 2. Attendance and apologies Present: Alan T. Hutton (President, ATH), Karl-Heinz Hellwich (Past-President, KHH), Risto S. Laitinen (Secretary, RSL), Michael A. Beckett (MAB), Edwin C. Constable (ECC), Ture Damhus (TD), Richard M. Hartshorn (RMH), Elisabeth Mansfield (EM), Gerry P. Moss (GPM), Michelle M. Rogers (MMR), Molly A. Strausbaugh (MAS), Clare Tovee (CT), Andrey Yerin (AY) (see also Appendix 1) Apologies: Fabio Aricò (FA), Maria Atanasova Petrova (MA), Neil Burford (NB), (JC), Ana Maria da Costa Ferreira (ACF), Safiye Erdem (SE), Rafał Kruszyński (RK), Robin Macaluso (RM), , Ladda Meesuk (LM), Ebbe Nordlander (EN), Amelia P. Rauter (APR), Erik Szabó (ES), Keith T. Taylor (KTT), Jiří Vohlídal (JV) Invited observer: G. Jeffery Leigh (GJL) No replies: József Nagy, Sangho Koo 3. Introduction of attendees A short round of introductions was made. A new titular member, Prof. Edwin C. Constable and a new Associate Member, Dr. Clare Tovee, were attending the meeting of the Division Committee for the first time in this function. KHH informed the meeting that the following persons had passed away during recent years: Peter A. -

(12) Patent Application Publication (10) Pub. No.: US 2005/0044778A1 Orr (43) Pub
US 20050044778A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2005/0044778A1 Orr (43) Pub. Date: Mar. 3, 2005 (54) FUEL COMPOSITIONS EMPLOYING Publication Classification CATALYST COMBUSTION STRUCTURE (51) Int. CI.' ........ C10L 1/28; C1OL 1/24; C1OL 1/18; (76) Inventor: William C. Orr, Denver, CO (US) C1OL 1/12; C1OL 1/26 Correspondence Address: (52) U.S. Cl. ................. 44/320; 44/435; 44/378; 44/388; HOGAN & HARTSON LLP 44/385; 44/444; 44/443 ONE TABOR CENTER, SUITE 1500 1200 SEVENTEENTH ST DENVER, CO 80202 (US) (57) ABSTRACT (21) Appl. No.: 10/722,127 Metallic vapor phase fuel compositions relating to a broad (22) Filed: Nov. 24, 2003 Spectrum of pollution reducing, improved combustion per Related U.S. Application Data formance, and enhanced Stability fuel compositions for use in jet, aviation, turbine, diesel, gasoline, and other combus (63) Continuation-in-part of application No. 08/986,891, tion applications include co-combustion agents preferably filed on Dec. 8, 1997, now Pat. No. 6,652,608. including trimethoxymethylsilane. Patent Application Publication Mar. 3, 2005 US 2005/0044778A1 FIGURE 1 CALCULATING BUNSEN BURNER LAMINAR FLAME VELOCITY (LFV) OR BURNING VELOCITY (BV) CONVENTIONAL FLAME LUMINOUS FLAME Method For Calculating Bunsen Burner Laminar Flame Velocity (LHV) or Burning Velocity Requires Inside Laminar Cone Angle (0) and The Gas Velocity (Vg). LFV = A, SIN 2 x VG US 2005/0044778A1 Mar. 3, 2005 FUEL COMPOSITIONS EMPLOYING CATALYST Chart of Elements (CAS version), and mixture, wherein said COMBUSTION STRUCTURE element or derivative compound, is combustible, and option 0001) The present invention is a CIP of my U.S. -

Operation Permit Application
Un; iy^\ tea 0 9 o Operation Permit Application Located at: 2002 North Orient Road Tampa, Florida 33619 (813) 623-5302 o Training Program TRAINING PROGRAM for Universal Waste & Transit Orient Road Tampa, Florida m ^^^^ HAZARDOUS WAb 1 P.ER^AlTTlNG TRAINING PROGRAM MASTER INDEX CHAPTER 1: Introduction Tab A CHAPTER 2: General Safety Manual Tab B CHAPTER 3: Protective Clothing Guide Tab C CHAPTER 4: Respiratory Training Program Tab D APPENDIX 1: Respiratory Training Program II Tab E CHAPTER 5: Basic Emergency Training Guide Tab F CHAPTER 6: Facility Operations Manual Tab G CHAPTER 7: Land Ban Certificates Tab H CHAPTER 8: Employee Certification Statement Tab. I CHAPTER ONE INTRODUCTION prepared by Universal Waste & Transit Orient Road Tampa Florida Introducti on STORAGE/TREATMENT PERSONNEL TRAINING PROGRAM All personnel involved in any handling, transportation, storage or treatment of hazardous wastes are required to start the enclosed training program within one-week after the initiation of employment at Universal Waste & Transit. This training program includes the following: Safety Equipment Personnel Protective Equipment First Aid & CPR Waste Handling Procedures Release Prevention & Response Decontamination Procedures Facility Operations Facility Maintenance Transportation Requirements Recordkeeping We highly recommend that all personnel involved in the handling, transportation, storage or treatment of hazardous wastes actively pursue additional technical courses at either the University of South Florida, or Tampa Junior College. Recommended courses would include general chemistry; analytical chemistry; environmental chemistry; toxicology; and additional safety and health related topics. Universal Waste & Transit will pay all registration, tuition and book fees for any courses which are job related. The only requirement is the successful completion of that course. -

UC Riverside Electronic Theses and Dissertations
UC Riverside UC Riverside Electronic Theses and Dissertations Title Carboranes: Building Blocks for Materials and Ligand Development Permalink https://escholarship.org/uc/item/2vp9m2z6 Author Estrada, Jess Steven Publication Date 2017 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA RIVERSIDE Carboranes: Building Blocks for Materials and Ligand Development A Dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Chemistry by Jess Steven Estrada September 2017 Dissertation Committee: Dr. Vincent Lavallo, Chairperson Dr. Richard Hooley Dr. Pingyun Feng Copyright by Jess Steven Estrada 2017 The Dissertation Jess Steven Estrada is approved: Committee Chairperson University of California, Riverside Acknowledgements I would like to thank Dr. Vincent Lavallo for giving me the opportunity to join his lab and for all of his help and support throughout graduate school. I owe a lot of my success to you. I would also like to thank the amazing faculty at UCR for all of their help and knowledge they have provided. I truly enjoyed every class I took here at UCR. In addition to the faculty, the staff in the chemistry department has been amazing over the last five years and played a huge role in my success as well so I would specifically like to thank Dr. Borchardt, the NMR genius, Dr. Fook for all of his help with my great looking X-ray structures, Christina Youhas for answering literally every question I ever had and being so kind about answering. My lab mates, who created the most unique group of people probably in the history of UCR and that I’ve ever had the pleasure of working with. -

WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C12N 15/10 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/DK20 15/050343 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 11 November 2015 ( 11. 1 1.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: PA 2014 00655 11 November 2014 ( 11. 1 1.2014) DK (84) Designated States (unless otherwise indicated, for every 62/077,933 11 November 2014 ( 11. 11.2014) US kind of regional protection available): ARIPO (BW, GH, 62/202,3 18 7 August 2015 (07.08.2015) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, (71) Applicant: LUNDORF PEDERSEN MATERIALS APS TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, [DK/DK]; Nordvej 16 B, Himmelev, DK-4000 Roskilde DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, (DK). -

Standard Thermodynamic Properties of Chemical
STANDARD THERMODYNAMIC PROPERTIES OF CHEMICAL SUBSTANCES ∆ ° –1 ∆ ° –1 ° –1 –1 –1 –1 Molecular fH /kJ mol fG /kJ mol S /J mol K Cp/J mol K formula Name Crys. Liq. Gas Crys. Liq. Gas Crys. Liq. Gas Crys. Liq. Gas Ac Actinium 0.0 406.0 366.0 56.5 188.1 27.2 20.8 Ag Silver 0.0 284.9 246.0 42.6 173.0 25.4 20.8 AgBr Silver(I) bromide -100.4 -96.9 107.1 52.4 AgBrO3 Silver(I) bromate -10.5 71.3 151.9 AgCl Silver(I) chloride -127.0 -109.8 96.3 50.8 AgClO3 Silver(I) chlorate -30.3 64.5 142.0 AgClO4 Silver(I) perchlorate -31.1 AgF Silver(I) fluoride -204.6 AgF2 Silver(II) fluoride -360.0 AgI Silver(I) iodide -61.8 -66.2 115.5 56.8 AgIO3 Silver(I) iodate -171.1 -93.7 149.4 102.9 AgNO3 Silver(I) nitrate -124.4 -33.4 140.9 93.1 Ag2 Disilver 410.0 358.8 257.1 37.0 Ag2CrO4 Silver(I) chromate -731.7 -641.8 217.6 142.3 Ag2O Silver(I) oxide -31.1 -11.2 121.3 65.9 Ag2O2 Silver(II) oxide -24.3 27.6 117.0 88.0 Ag2O3 Silver(III) oxide 33.9 121.4 100.0 Ag2O4S Silver(I) sulfate -715.9 -618.4 200.4 131.4 Ag2S Silver(I) sulfide (argentite) -32.6 -40.7 144.0 76.5 Al Aluminum 0.0 330.0 289.4 28.3 164.6 24.4 21.4 AlB3H12 Aluminum borohydride -16.3 13.0 145.0 147.0 289.1 379.2 194.6 AlBr Aluminum monobromide -4.0 -42.0 239.5 35.6 AlBr3 Aluminum tribromide -527.2 -425.1 180.2 100.6 AlCl Aluminum monochloride -47.7 -74.1 228.1 35.0 AlCl2 Aluminum dichloride -331.0 AlCl3 Aluminum trichloride -704.2 -583.2 -628.8 109.3 91.1 AlF Aluminum monofluoride -258.2 -283.7 215.0 31.9 AlF3 Aluminum trifluoride -1510.4 -1204.6 -1431.1 -1188.2 66.5 277.1 75.1 62.6 AlF4Na Sodium tetrafluoroaluminate -

Sop Pyrophoric 2 12/16/2019
Owner DOC. NO. REV. DATE C.H.O SOP PYROPHORIC 2 12/16/2019 DOC. TITLE SOP FOR PYROPHORIC CHEMICALS Environmental Health & Safety STANDARD OPERATING PROCEDURES (SOP) FOR WORKING WITH PYROPHORIC CHEMICALS AT AMHERST COLLEGE ___________________________________________________________________ General Information Pyrophoric Chemicals are solid, liquid, or gas compounds that, when exposed to air or moisture at or below 54°C (130°F), can spontaneously ignite. Examples of Pyrophoric chemicals used at Amherst College Laboratories include: sodium hydride, zinc powder, and Grignard reagents. See the “Appendix” page below for a full list of Pyrophoric Chemicals. Pyrophoric chemicals are often used as catalysts in chemical reactions or as reducing and deprotonating agents in organic chemistry. Note that Pyrophoric chemicals may also be characterized by other hazards, hence, users of these chemicals may also need to refer to other SOPs that cover other hazards. In addition, each individual chemical’s Safety Data Sheet (SDS) should be consulted before they are used. _____________________________________________________________________________________ Personal Protective Equipment When working with Pyrophoric Chemicals, the following personal protective equipment (PPE) must be worn, at a minimum. Depending on the specific chemical, other forms of protection might be required. Consult the SDS for each chemical before use: Splash goggles Lab coat (Fire resistant lab coat highly recommended) Long pants Close toed shoes Gloves – Nitrile gloves adequate for accidental contact with small quantities. However, the use of fire resistant Nomex/ Leather Pilot’s gloves is highly recommended _____________________________________________________________________________________ Safety Devices All work with Pyrophoric chemicals must be done in a glove box, vacuum manifold, or any enclosed inert environment. If work must be done in a fume hood, ensure that the hood sash is in the lowest feasible position. -

United States Patent Office Patented Jan
3,489,812 United States Patent Office Patented Jan. 13, 1970 2 3,489,812 and sufficient dialkyl sulfide to permit convenient stirring METHOD OF PREPARING BIS-(DIETHYL of the reactant mixture. The reaction should be carried SULFIDE)-DECABORANE out in the absence of air or other oxidizing gas, suitably Mervin D. Marshall, Fombell, and Richard M. Hunt and by using a vacuum or nonoxidizing cover gas such as Gerald T. Heiferan, Butler, Pa., assignors to Mine nitrogen or argon. Safety Appliances Company, a corporation of Pennsyl vania tion:The following examples are illustrative of this inven No Drawing. Filed Jan. 15, 1968, Ser. No. 697,596 Int, C. C07f 5/02 EXAMPLE I U.S. C. 260-606.5 1 Claim O A glass reactor equipped with a magnetic stirrer and connected to a glass vacuum apparatus was charged with ABSTRACT OF THE DISCLOSURE 3.513 g. of (NH4)2BioHo (22.6 millimols) and 40 ml. of diethyl sulfide, cooled to about 4° C. and evacuated. Bis-(diethyl sulfide)-decaborane is prepared by reac Measured aliquots of anhydrous hydrogen chloride (92. tion of ammonium decahydrodecarborate, HCl and di 15 immoles) were added in two equal increments. This reac ethyl sulfide. tion mixture was allowed to warm to room temperature and stirred for 72 hours. A white precipitate formed which Bis-(dialkyl sulfide)-decaboranes, such as bis-(dimethyl was filtered from the diethyl sulfide solution. The evapora sulfide)-decaborane, BioH12S (CH3)2)2, and bis-(diethyl tion of excess diethyl sulfide from the solution gave 4.596 sulfide)-decaborane, BioH12S (C2H5)2)2, have been used g. -

Nite States Patent O?Ice Patented Apr
3,030,423 nite States Patent O?ice Patented Apr. 17, 1962 1 2 cuprous chloride catalyst employed also can be varied 3,030,423 REACTION PRODUCTS 0F ACETYLENES widely, generally being in the range of from 0.005 to 0.5 AND DECABORANE mole of cuprous chloride per mole of decaborane and Starling K. Alley, Beverly Hills, Calif., and Otto Fuchs, preferably from 0.01 to 0.2 mole of cuprous chloride per Tonawanzia, N.Y., assignors to Olin Mathieson Chemi- 5 mole of decaborane. The reaction temperature can vary cal Corporation, a corporation of Virginia from 100 to 350° C. and preferably from 150 to 250° C. No Drawing. Filed Feb. 13, 1959, Ser. No. 793,211 The pressure can vary from atmospheric pressure to about 3 Claims. (Cl. 260-6065) 700 p.s.i.g., although closed system reactions are usually conducted at about 100 to 600 p.s.i.g. The reaction gen This invention relates to solid products obtained by 10 erally requires about 1 to 10 hours depending upon the the reaction of acetylene or methylacetylene with deca~ ratio of reactants and the temperature and pressure em borane. ployed. The solid products of this invention, when incorporated Although the reaction will proceed in the absence of a with suitable oxidizers such as ammonium perchlorate, solvent, a solvent common for the reactants but inert with potassium perchlorate, sodium perchlorate, etc., yield 15 respect to the reactants under the reaction conditions is solid propellants generally suitable for rocket power usually employed. Such solvents include aliphatic hydro plants and other jet propelled devices. -

RU041 0001 Al
RU041 0001 A i I Aj I Tol; I Al DISCLAIMER Images are produced from the best available original document, nevertheless portions of this document may be illegible in electronic image products. Disclaimer Please be aware that all missing pages were supposed to be blank. ORGANIZERS Russian Academy of Sciences (RAS) Division of Cemistry ad Material Sciences Researc Cuncil Oil Organic ad Or-anoelenient Cenlistly AX Nesrnevario, Institute of Organoelerrient Compounds N.D. elinsky Institute of Organic Chemistry N.S. Kurnakm Istitute of General and Inorganic Cemistry Institute for Physical Chemistry of Ceramics M.V. Lomoiiosov iMoscow State University St"ite ScIcimfic-Research Istitute of Cliernisti- ad Technology of Organoelerrient Compounds ,,VIAM, State Research Center SPONSORS I U PAC U N ESCO Ministry of Industry, Science ad Technology of te Russian Federation RLISSIaii Foundation for Basic Research BORAX Europe Ltd. AV!;'WOR OAO WELCOME TO IMEBORON XI Dear Colleazues, Welcome to the XIth International Conference onBoron Chemistry in Moscow. Boron Chemistry as a connecting bridge between many fields aintains one of the leading positions in modem chemistry. This is reflected both y an ncreasing number of publications on organoboron, carborane and metallacarborane chemistry and a lot of boron conferences of International, European and National formats. We feel very much honored that the MEBORON XI, the first Iternational Conference on Boron Chemistry in the XXI Century, will be held in Moscow, te Capital of the Russian Federation. This Moscow Conference will take place in the conference halls (the Red and Blue oes) of te main building Of th Rsian Academy of Sciences, 32a, Lemnsky prospect, 3 d floor.