UC Riverside Electronic Theses and Dissertations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
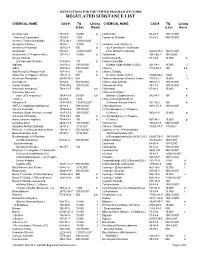
Regulated Substance List
INSTRUCTIONS FOR THE UNIFIED PROGRAM (UP) FORM REGULATED SUBSTANCE LIST CHEMICAL NAME CAS # TQ Listing CHEMICAL NAME CAS # TQ Listing (Lbs) Basis (Lbs) Basis Acetaldehyde 75-07-0 10,000 g Cantharidin 56-25-7 100/10,0001 * Acetone Cyanohydrin 75-86-5 1,000 Carbachol Chloride 51-83-2 500/10,0001 Acetone Thiosemicarbazide 1752-30-3 1,000/10,0001 Acetylene (Ethyne) 74-86-2 10,000 f Carbamic Acid, Methyl-,o- Acrolein (2-Propenal) 107-02-8 500 b (((2,4-Dimethyl-1,3-Dithiolan- Acrylamide 79-06-1 1,000/10,0001 2-YL) Methylene)Amino)- 26419-73-8 100/10,0001 Acrylonitrile (2- Propenenitrile) 107-13-1 10,000 b Carbofuran 1563-66-2 10/10,0001 Acrylyl Chloride Carbon Disulfide 75-15-0 10,000 b (2-Propenoyl Chloride) 814-68-6 100 b Carbon Oxysulfide Aldicarb 116-06-3 100/10,0001 (Carbon Oxide Sulfide (COS)) 463-58-1 10,000 f Aldrin 309-00-2 500/10,0001 Chlorine 7782-50-5 100 a,b Allyl Alcohol (2-Propen-1-ol) 107-18-6 1,000 b Chlorine Dioxide Allylamine (2-Propen-1-Amine) 107-11-9 500 b (Chlorine Oxide (ClO2)) 10049-04-4 1,000 c Aluminum Phosphide 20859-73-8 500 Chlorine Monoxide (Chlorine Oxide) 7791-21-1 10,000 f Aminopterin 54-62-6 500/10,0001 Chlormequat Chloride 999-81-5 100/10,0001 Amiton Oxalate 3734-97-2 100/10,0001 Chloroacetic Acid 79-11-8 100/10,0001 Ammonia, Anhydrous 2 7664-41-7 500 a,b Chloroform 67-66-3 10,000 b Ammonia, Aqueous Chloromethyl Ether (conc 20% or greater) 7664-41-7 20,000 a,b (Methane,Oxybis(chloro-) 542-88-1 100 b * Aniline 62-53-3 1,000 Chloromethyl Methyl Ether Antimycin A 1397-94-0 1,000/10,0001 (Chloromethoxymethane) -

Thermodynamic Hydricity of Small Borane Clusters and Polyhedral Closo-Boranes
molecules Article Thermodynamic Hydricity of Small Borane Clusters y and Polyhedral closo-Boranes Igor E. Golub 1,* , Oleg A. Filippov 1 , Vasilisa A. Kulikova 1,2, Natalia V. Belkova 1 , Lina M. Epstein 1 and Elena S. Shubina 1,* 1 A. N. Nesmeyanov Institute of Organoelement Compounds and Russian Academy of Sciences (INEOS RAS), 28 Vavilova St, 119991 Moscow, Russia; [email protected] (O.A.F.); [email protected] (V.A.K.); [email protected] (N.V.B.); [email protected] (L.M.E.) 2 Faculty of Chemistry, M.V. Lomonosov Moscow State University, 1/3 Leninskiye Gory, 119991 Moscow, Russia * Correspondence: [email protected] (I.E.G.); [email protected] (E.S.S.) Dedicated to Professor Bohumil Štibr (1940-2020), who unfortunately passed away before he could reach the y age of 80, in the recognition of his outstanding contributions to boron chemistry. Academic Editors: Igor B. Sivaev, Narayan S. Hosmane and Bohumír Gr˝uner Received: 6 June 2020; Accepted: 23 June 2020; Published: 25 June 2020 MeCN Abstract: Thermodynamic hydricity (HDA ) determined as Gibbs free energy (DG◦[H]−) of the H− detachment reaction in acetonitrile (MeCN) was assessed for 144 small borane clusters (up 2 to 5 boron atoms), polyhedral closo-boranes dianions [BnHn] −, and their lithium salts Li2[BnHn] (n = 5–17) by DFT method [M06/6-311++G(d,p)] taking into account non-specific solvent effect (SMD MeCN model). Thermodynamic hydricity values of diborane B2H6 (HDA = 82.1 kcal/mol) and its 2 MeCN dianion [B2H6] − (HDA = 40.9 kcal/mol for Li2[B2H6]) can be selected as border points for the range of borane clusters’ reactivity. -

Minutesbasel Approved
Approved minutes, Basel 2018 International Union of Pure and Applied Chemistry Division VIII Chemical Nomenclature and Structure Representation Approved minutes for Division Committee Meeting Basel, Switzerland, 13–14 August, 2018 1. Welcome, introductory remarks and housekeeping announcements Alan Hutton (ATH) welcomed everybody to the meeting, extending a special welcome to those who were attending the Division Committee (DC) meeting for the first time, and noting the presence of three former Presidents of the Division, as well as the current Secretary General of IUPAC. He described the house rules and arrangements for the course of the meeting. 2. Attendance and apologies Present: Alan T. Hutton (President, ATH), Karl-Heinz Hellwich (Past-President, KHH), Risto S. Laitinen (Secretary, RSL), Michael A. Beckett (MAB), Edwin C. Constable (ECC), Ture Damhus (TD), Richard M. Hartshorn (RMH), Elisabeth Mansfield (EM), Gerry P. Moss (GPM), Michelle M. Rogers (MMR), Molly A. Strausbaugh (MAS), Clare Tovee (CT), Andrey Yerin (AY) (see also Appendix 1) Apologies: Fabio Aricò (FA), Maria Atanasova Petrova (MA), Neil Burford (NB), (JC), Ana Maria da Costa Ferreira (ACF), Safiye Erdem (SE), Rafał Kruszyński (RK), Robin Macaluso (RM), , Ladda Meesuk (LM), Ebbe Nordlander (EN), Amelia P. Rauter (APR), Erik Szabó (ES), Keith T. Taylor (KTT), Jiří Vohlídal (JV) Invited observer: G. Jeffery Leigh (GJL) No replies: József Nagy, Sangho Koo 3. Introduction of attendees A short round of introductions was made. A new titular member, Prof. Edwin C. Constable and a new Associate Member, Dr. Clare Tovee, were attending the meeting of the Division Committee for the first time in this function. KHH informed the meeting that the following persons had passed away during recent years: Peter A. -

United States Patent Office Patented Jan
3,489,812 United States Patent Office Patented Jan. 13, 1970 2 3,489,812 and sufficient dialkyl sulfide to permit convenient stirring METHOD OF PREPARING BIS-(DIETHYL of the reactant mixture. The reaction should be carried SULFIDE)-DECABORANE out in the absence of air or other oxidizing gas, suitably Mervin D. Marshall, Fombell, and Richard M. Hunt and by using a vacuum or nonoxidizing cover gas such as Gerald T. Heiferan, Butler, Pa., assignors to Mine nitrogen or argon. Safety Appliances Company, a corporation of Pennsyl vania tion:The following examples are illustrative of this inven No Drawing. Filed Jan. 15, 1968, Ser. No. 697,596 Int, C. C07f 5/02 EXAMPLE I U.S. C. 260-606.5 1 Claim O A glass reactor equipped with a magnetic stirrer and connected to a glass vacuum apparatus was charged with ABSTRACT OF THE DISCLOSURE 3.513 g. of (NH4)2BioHo (22.6 millimols) and 40 ml. of diethyl sulfide, cooled to about 4° C. and evacuated. Bis-(diethyl sulfide)-decaborane is prepared by reac Measured aliquots of anhydrous hydrogen chloride (92. tion of ammonium decahydrodecarborate, HCl and di 15 immoles) were added in two equal increments. This reac ethyl sulfide. tion mixture was allowed to warm to room temperature and stirred for 72 hours. A white precipitate formed which Bis-(dialkyl sulfide)-decaboranes, such as bis-(dimethyl was filtered from the diethyl sulfide solution. The evapora sulfide)-decaborane, BioH12S (CH3)2)2, and bis-(diethyl tion of excess diethyl sulfide from the solution gave 4.596 sulfide)-decaborane, BioH12S (C2H5)2)2, have been used g. -

Nite States Patent O?Ice Patented Apr
3,030,423 nite States Patent O?ice Patented Apr. 17, 1962 1 2 cuprous chloride catalyst employed also can be varied 3,030,423 REACTION PRODUCTS 0F ACETYLENES widely, generally being in the range of from 0.005 to 0.5 AND DECABORANE mole of cuprous chloride per mole of decaborane and Starling K. Alley, Beverly Hills, Calif., and Otto Fuchs, preferably from 0.01 to 0.2 mole of cuprous chloride per Tonawanzia, N.Y., assignors to Olin Mathieson Chemi- 5 mole of decaborane. The reaction temperature can vary cal Corporation, a corporation of Virginia from 100 to 350° C. and preferably from 150 to 250° C. No Drawing. Filed Feb. 13, 1959, Ser. No. 793,211 The pressure can vary from atmospheric pressure to about 3 Claims. (Cl. 260-6065) 700 p.s.i.g., although closed system reactions are usually conducted at about 100 to 600 p.s.i.g. The reaction gen This invention relates to solid products obtained by 10 erally requires about 1 to 10 hours depending upon the the reaction of acetylene or methylacetylene with deca~ ratio of reactants and the temperature and pressure em borane. ployed. The solid products of this invention, when incorporated Although the reaction will proceed in the absence of a with suitable oxidizers such as ammonium perchlorate, solvent, a solvent common for the reactants but inert with potassium perchlorate, sodium perchlorate, etc., yield 15 respect to the reactants under the reaction conditions is solid propellants generally suitable for rocket power usually employed. Such solvents include aliphatic hydro plants and other jet propelled devices. -

The Use of Carboranes in Cancer Drug Develop-Ment
ISSN: 2378-3419 Zargham et al. Int J Cancer Clin Res 2019, 6:110 DOI: 10.23937/2378-3419/1410110 Volume 6 | Issue 2 International Journal of Open Access Cancer and Clinical Research REviEW ARtiCLE The Use of Carboranes in Cancer Drug Development Emilia O Zargham, Christian A Mason and Mark W Lee Jr* Check for Department of Chemistry, University of Missouri, Columbia, Missouri, USA updates *Corresponding author: Mark W Lee Jr., Department of Chemistry, University of Missouri, 601 South College Avenue, Columbia, Missouri, 65211, USA, Tel: 573-884-2424 erwise rapidly metabolize [5]. Furthermore, selective Abstract chemical substitution of each carbon or boron atom Over the past decade, there has been a rising interest in these clusters allows for their use as rigid, three di- in the use of carboranes as a potential pharmacophoric moiety in the development of new drugs for the treatment of mensional scaffolds upon which to construct new drug various types of cancer. The unique physical and chemical molecules. properties of carboranes make their use attractive in drug development. In several instances, the inclusion of Nearly all past biomedical research involving carbo- carboranes into a drug structure has increased the agent’s ranes has focused on their use in the design of boron de- binding affinity, potency, or bioavailability. The purpose of livery agents for boron neutron capture therapy (BNCT) this review is to highlight applications of carboranes to the [2]. This binary radiation therapy depends on the selec- medicinal chemistry of cancer. tive delivery of a high concentration of boron-10 atoms to targeted tissues. -

Carborane Derivatives with Electron Rich Moieties. Synthesis, Properties and Electronic
Carborane derivatives with electron rich moieties. Synthesis, properties and electronic communication. RADU-ADRIAN POPESCU TESI DOCTORAL Programa de Doctorat en Química Directora: Prof. Clara Viñas Teixidor Departament de Química Facultat de Ciències 2012 Memòria presentada per aspirar al Grau de Doctor per Radu‐Adrian Popescu Vist i plau Prof. Clara Viñas Teixidor Bellaterra, 16 de novembre de 2012 La Professora CLARA VIÑAS i TEIXIDOR, Professora d’Investigació del Consejo Superior de Investigaciones Científicas a l’Institut de Ciència de Materials de Barcelona CERTIFICA Que en RADU‐ADRIAN POPESCU, llicenciat en Enginyeria Química, ha realitzat sota la meva direcció la Tesí Doctoral que porta per títol “Carborane derivatives with electron rich moieties. Synthesis, properties and electronic communication” i que recull aquesta memòria per optar al títol de Doctor en Química per la Universitat Autònoma de Barcelona. I, perquè consti i tingui els efectes corresponents, signa aquest certificat a Bellaterra, a 16 de novembre de 2012. Prof. CLARA VIÑAS i TEIXIDOR ICMAB (CSIC) http://www.icmab.es Campus de la Universitat Autònoma de Barcelona 08193 Bellaterra, Catalunya, Espanya Telf.: +34 935 801 853 Fax.: +34 935 805 729 Aquest treball de recerca ha estat finançat per la Comisión Interministerial de Ciencia y Tecnología, CICYT, mitjançant el projecte CTQ2010-16237 (subprograma BQU) i per la Generalitat de Catalunya amb el projecte 2009/SGR/00279. Alhora, s’ha pogut realitzar gràcies a una beca per la Formació de Personal Universitari (FPU) concedida pel Miniesterio de Ciencia e Innovación, des del juliol del 2008 al juliol del 2012. Aquest treball d’investigació, amb la data de defensa del 18 de gener de 2013 , té com a membres del tribunal a: - Prof. -

Safety Data Sheet
SAFETY DATA SHEET Revision Date 14-February-2020 Revision Number 2 1. Identification Product Name Decaborane Cat No. : 87892 CAS-No 17702-41-9 Synonyms No information available Recommended Use Laboratory chemicals. Uses advised against Food, drug, pesticide or biocidal product use. Details of the supplier of the safety data sheet Company Importer/Distributor Manufacturer Fisher Scientific Alfa Aesar 112 Colonnade Road, Thermo Fisher Scientific Chemicals, Inc. Ottawa, ON K2E 7L6, 30 Bond Street, Ward Hill, MA 01835-8099 Canada Tel: 800-343-0660 Fax: 800-322-4757 Tel: 1-800-234-7437 Email: [email protected] www.alfa.com Emergency Telephone Number During normal business hours (Monday-Friday, 8am-7pm EST), call (800) 343-0660. After normal business hours, call Carechem 24 at (800) 579-7421. 2. Hazard(s) identification Classification WHMIS 2015 Classification Classified as hazardous under the Hazardous Products Regulations (SOR/2015-17) Flammable solids Category 2 Acute oral toxicity Category 3 Acute dermal toxicity Category 3 Acute Inhalation Toxicity Category 2 Physical Hazards Not Otherwise Classified Category 1 Risk of explosion if heated under confinement Label Elements Signal Word Danger Hazard Statements Flammable solid Fatal if inhaled Toxic if swallowed or in contact with skin ______________________________________________________________________________________________ Page 1 / 7 Decaborane Revision Date 14-February-2020 ______________________________________________________________________________________________ Risk of explosion if heated under confinement Precautionary Statements Manufacturer Alfa Aesar Thermo Fisher Scientific Chemicals, Inc. 30 Bond Street, Ward Hill, MA 01835-8099 Tel: 800-343-0660 Fax: 800-322-4757 Email: [email protected] www.alfa.com Prevention Keep away from heat, hot surfaces, sparks, open flames and other ignition sources. -

Tuning Photophysical Properties in Closo-Decaborane Cluster Derivatives
TUNING PHOTOPHYSICAL PROPERTIES IN CLOSO-DECABORANE CLUSTER DERIVATIVES by Mustapha B. Abdulmojeed A Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science in Chemistry Middle Tennessee State University 2020 Thesis Committee: Dr. Andrienne C. Friedli, Major Advisor Dr. Piotr Kaszynski, Committee Member Dr. Kevin Bicker, Committee Member Dr. Justin Miller, Committee Member ii Tuning Photophysical Properties in Closo-Decaborane Cluster Derivatives by Mustapha B. Abdulmojeed Approved: _____________________________________________ Dr. Andrienne C. Friedli, Committee Chair _____________________________________________ Dr. Piotr Kaszynski, Committee Member _____________________________________________ Dr. Kevin L. Bicker, Committee Member _____________________________________________ Dr. Justin M. Miller, Committee Member _____________________________________________ Dr. Paul G. Van Patten, Chair, Department of Chemistry _____________________________________________ Dr. David L. Butler, Dean, College of Graduate Studies ii iii ACKNOWLEDGEMENTS All thanks belong to God who has made it possible to complete this piece of work. Thanks to my wife for her love, care and understanding, taking care of our kids while I was away. Special thanks to my advisor, Dr. Andrienne Friedli for her support, guide and patiently delivered instructions through-out the course of this project. Dziękuję to my co- advisor, Dr. Piotr Kaszynski whose constant push and motivation from across the Atlantic Ocean kept me going, no amount of words could express my gratitude to both of you. Sincere appreciation also goes to Dr. Szymon Kapuscinski who is not just an excellent senior colleague but a great personality whose help in this work cannot be measured. I also want to thank MTSU chemistry department for the funding opportunity. I extend my gratitude to all faculty members in the department for creating a conducive and welcoming environment to learn and train as a chemist. -

Towards Carborane-Functionalized Structures for the Treatment of Brain Cancer
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Portsmouth University Research Portal (Pure) Towards carborane-functionalized structures for the treatment of brain cancer Authors: Gianpiero Calabresea (corresponding author), Anis Daoua, Eugen Barbub, John Tsibouklisb Affiliations: aSchool of Life Science, Pharmacy and Chemistry, Kingston University London, Penrhyn Road, Kingston-upon-Thames, Surrey, KT1 2EE, UK; bSchool of Pharmacy and Biomedical Sciences, University of Portsmouth, Portsmouth, PO1 2DT, UK. Corresponding author: Calabrese, G. ([email protected]); tel.: 020 8417 9000 Keywords: BNCT, carboranes, brain cancer, boronated agents, clinical efficacy. Teaser: The development of carborane derivatives with high cancer-cell targeting specificity is key to these materials fulfilling their promise as the clinical BNCT agents of the future. Author biography See attached file Abstract Boron Neutron Capture Therapy (BNCT) is a promising targeted chemo-radiotherapeutic technique for the management of invasive brain tumours, such as glioblastoma multiforme. Prerequisite to effective BNCT is the selective targeting of tumour cells with 10B-rich therapeutic moieties. To this end, polyhedral boranes, especially carboranes, have received considerable attention since they combine high boron content, relative low toxicity and metabolic inertness. This article reviews progresses in the molecular design of recently investigated carborane derivatives in light of the widely accepted performance requirements for effective BNCT. 2 The principles of Boron Neutron Capture Therapy BNCT is a two-step targeted chemo-radiotherapeutic technique that involves the selective delivery of 10B-rich agents to cancer cells for the purpose of their selective destruction through subsequent irradiation with low- energy neutrons, which initiate highly localised nuclear fission reactions that do not damage surrounding tissue (Figure 1). -

And 1,7-Carborane Ligands for Boron Neutron Capture Therapy
YO t' Lg-3^o Ptatinum(Il) Complexes Containing lr2' and lr7-Carborane Ligands for Boron Neutron Capture Therapy A Thesis Submitted Towards the Degree of Doctor ofPhilosophy by Jean Ann Todd B.Sc. (Hons) * * a ADELAIDE * t UNIVERSITY t- AUSTRALIA cRucr L Department of Chemistry October 2001 Contents Acknowledgements IX Statement x Abstract xl Abbreviations xiii Chapter I : Introduction I l.l Cancer 1.1.1 Current Cancer Therapy 2 1.1.2 Binary Systems 2 l.l.2.l The Neutron Capture Reaction 2 1.2 Boron Neutron Capture Therapy J 1.2.1 The Advantages of BNCT J 1.2.2 Optimisation of BNCT 4 1.2.3 Application of BNCT 5 1.2.3.1 The Blood-Brain Barrier 5 1.2.4 Early Trials of BNCT 6 1.2.5 Recent Advances in BNCT 8 1.2.5.1The Carboranes 8 l.2.5.2Types of BNCT Agents 9 1.3 Tumour Selective Agents l0 1.3.1 Cellular Builcling Blocks l0 I 1.3.1. I Boron-Containing Nucleic Acid Precursors I .3.1.2Boron-Containing Amino Acids 1.3.1.3 Lipids, Phospholipids and Carbohydrates 1.3.2 Lipoproteins 1.3.3 Liposomes 1.3.4 Receptor / Antigen Binders 1.3.4.1Antibodies l-3.4.2Growth Factors 1.3.4.3 Hormones 1.3.5 Porphyrins 1.4 DNA Targeting Agents for BNCT 1.4.1 AlkylatingAgents L4.2 DNA Intercalators 1.4.3 Groove Binders I.4.4 Polyamines 1.4.5 Nucleic Acid Precursors 1.5 Background of platinum anti-cancer drugs 1.5.1 Cisplatin 1.5.1 .1 Activity and Action of Cisplatin 1.5.1.2 Problems Associated with the Use of Cisplatin in Cancer Therapy 1.5.2 New Generations of Platinum Anti-tumour Complexes 1.5.2.1Carboplatin 1.5.2.2 Other analogues of Cisplatin and Carboplatin- -

And 5-Halodecaboranes: Selective Syntheses from Closo-B10H10(2-) and Use As Polyborane Building Blocks" (2010)
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations Fall 2010 6- and 5-Halodecaboranes: Selective Syntheses From ClOSO- B10H10(2-) and Use as Polyborane Building Blocks William C. Ewing University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Inorganic Chemistry Commons Recommended Citation Ewing, William C., "6- and 5-Halodecaboranes: Selective Syntheses From ClOSO-B10H10(2-) and Use as Polyborane Building Blocks" (2010). Publicly Accessible Penn Dissertations. 261. https://repository.upenn.edu/edissertations/261 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/261 For more information, please contact [email protected]. 6- and 5-Halodecaboranes: Selective Syntheses From ClOSO-B10H10(2-) and Use as Polyborane Building Blocks Abstract Decaborane halogenated in the 6-position has been synthesized in high yields via the super-acid induced cage-opening reactions of closo-B10H10(2-) salts. These 6-halogenated compounds were then isomerized to their 5-substituted isomers through base catalysis. The isomerization was driven by the energy differences between the anionic-forms of each respective isomer. These reactions provided 5-halodeboranes in high yields. The bridging-hydrogens of the halodecaboranyl anions were fluxional at a range of temperatures. Variable-temperature NMR studies supported computationally proposed fluxional mechanisms. Both 5- and 6-halodecaboranes were reacted with alcohols yielding boranyl ethers. The mechanisms of substitution, where reactions with 6- and 5-halodecaboranes yielded 5- and 6-boranyl ethers, respectively, were explained computationally and confirmed through isotopic-labeling studies. The regeneration of the polymeric products of ammonia-borane dehydrogenation was carried out through a process that included digestion of the polymer, complexation of the digestate with a base, reduction of B-X bonds to B-H bonds, and finally displacement of the base with ammonia.