And 5-Halodecaboranes: Selective Syntheses from Closo-B10H10(2-) and Use As Polyborane Building Blocks" (2010)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
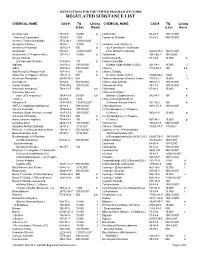
Regulated Substance List
INSTRUCTIONS FOR THE UNIFIED PROGRAM (UP) FORM REGULATED SUBSTANCE LIST CHEMICAL NAME CAS # TQ Listing CHEMICAL NAME CAS # TQ Listing (Lbs) Basis (Lbs) Basis Acetaldehyde 75-07-0 10,000 g Cantharidin 56-25-7 100/10,0001 * Acetone Cyanohydrin 75-86-5 1,000 Carbachol Chloride 51-83-2 500/10,0001 Acetone Thiosemicarbazide 1752-30-3 1,000/10,0001 Acetylene (Ethyne) 74-86-2 10,000 f Carbamic Acid, Methyl-,o- Acrolein (2-Propenal) 107-02-8 500 b (((2,4-Dimethyl-1,3-Dithiolan- Acrylamide 79-06-1 1,000/10,0001 2-YL) Methylene)Amino)- 26419-73-8 100/10,0001 Acrylonitrile (2- Propenenitrile) 107-13-1 10,000 b Carbofuran 1563-66-2 10/10,0001 Acrylyl Chloride Carbon Disulfide 75-15-0 10,000 b (2-Propenoyl Chloride) 814-68-6 100 b Carbon Oxysulfide Aldicarb 116-06-3 100/10,0001 (Carbon Oxide Sulfide (COS)) 463-58-1 10,000 f Aldrin 309-00-2 500/10,0001 Chlorine 7782-50-5 100 a,b Allyl Alcohol (2-Propen-1-ol) 107-18-6 1,000 b Chlorine Dioxide Allylamine (2-Propen-1-Amine) 107-11-9 500 b (Chlorine Oxide (ClO2)) 10049-04-4 1,000 c Aluminum Phosphide 20859-73-8 500 Chlorine Monoxide (Chlorine Oxide) 7791-21-1 10,000 f Aminopterin 54-62-6 500/10,0001 Chlormequat Chloride 999-81-5 100/10,0001 Amiton Oxalate 3734-97-2 100/10,0001 Chloroacetic Acid 79-11-8 100/10,0001 Ammonia, Anhydrous 2 7664-41-7 500 a,b Chloroform 67-66-3 10,000 b Ammonia, Aqueous Chloromethyl Ether (conc 20% or greater) 7664-41-7 20,000 a,b (Methane,Oxybis(chloro-) 542-88-1 100 b * Aniline 62-53-3 1,000 Chloromethyl Methyl Ether Antimycin A 1397-94-0 1,000/10,0001 (Chloromethoxymethane) -

Thermodynamic Hydricity of Small Borane Clusters and Polyhedral Closo-Boranes
molecules Article Thermodynamic Hydricity of Small Borane Clusters y and Polyhedral closo-Boranes Igor E. Golub 1,* , Oleg A. Filippov 1 , Vasilisa A. Kulikova 1,2, Natalia V. Belkova 1 , Lina M. Epstein 1 and Elena S. Shubina 1,* 1 A. N. Nesmeyanov Institute of Organoelement Compounds and Russian Academy of Sciences (INEOS RAS), 28 Vavilova St, 119991 Moscow, Russia; [email protected] (O.A.F.); [email protected] (V.A.K.); [email protected] (N.V.B.); [email protected] (L.M.E.) 2 Faculty of Chemistry, M.V. Lomonosov Moscow State University, 1/3 Leninskiye Gory, 119991 Moscow, Russia * Correspondence: [email protected] (I.E.G.); [email protected] (E.S.S.) Dedicated to Professor Bohumil Štibr (1940-2020), who unfortunately passed away before he could reach the y age of 80, in the recognition of his outstanding contributions to boron chemistry. Academic Editors: Igor B. Sivaev, Narayan S. Hosmane and Bohumír Gr˝uner Received: 6 June 2020; Accepted: 23 June 2020; Published: 25 June 2020 MeCN Abstract: Thermodynamic hydricity (HDA ) determined as Gibbs free energy (DG◦[H]−) of the H− detachment reaction in acetonitrile (MeCN) was assessed for 144 small borane clusters (up 2 to 5 boron atoms), polyhedral closo-boranes dianions [BnHn] −, and their lithium salts Li2[BnHn] (n = 5–17) by DFT method [M06/6-311++G(d,p)] taking into account non-specific solvent effect (SMD MeCN model). Thermodynamic hydricity values of diborane B2H6 (HDA = 82.1 kcal/mol) and its 2 MeCN dianion [B2H6] − (HDA = 40.9 kcal/mol for Li2[B2H6]) can be selected as border points for the range of borane clusters’ reactivity. -

Minutesbasel Approved
Approved minutes, Basel 2018 International Union of Pure and Applied Chemistry Division VIII Chemical Nomenclature and Structure Representation Approved minutes for Division Committee Meeting Basel, Switzerland, 13–14 August, 2018 1. Welcome, introductory remarks and housekeeping announcements Alan Hutton (ATH) welcomed everybody to the meeting, extending a special welcome to those who were attending the Division Committee (DC) meeting for the first time, and noting the presence of three former Presidents of the Division, as well as the current Secretary General of IUPAC. He described the house rules and arrangements for the course of the meeting. 2. Attendance and apologies Present: Alan T. Hutton (President, ATH), Karl-Heinz Hellwich (Past-President, KHH), Risto S. Laitinen (Secretary, RSL), Michael A. Beckett (MAB), Edwin C. Constable (ECC), Ture Damhus (TD), Richard M. Hartshorn (RMH), Elisabeth Mansfield (EM), Gerry P. Moss (GPM), Michelle M. Rogers (MMR), Molly A. Strausbaugh (MAS), Clare Tovee (CT), Andrey Yerin (AY) (see also Appendix 1) Apologies: Fabio Aricò (FA), Maria Atanasova Petrova (MA), Neil Burford (NB), (JC), Ana Maria da Costa Ferreira (ACF), Safiye Erdem (SE), Rafał Kruszyński (RK), Robin Macaluso (RM), , Ladda Meesuk (LM), Ebbe Nordlander (EN), Amelia P. Rauter (APR), Erik Szabó (ES), Keith T. Taylor (KTT), Jiří Vohlídal (JV) Invited observer: G. Jeffery Leigh (GJL) No replies: József Nagy, Sangho Koo 3. Introduction of attendees A short round of introductions was made. A new titular member, Prof. Edwin C. Constable and a new Associate Member, Dr. Clare Tovee, were attending the meeting of the Division Committee for the first time in this function. KHH informed the meeting that the following persons had passed away during recent years: Peter A. -

UC Riverside Electronic Theses and Dissertations
UC Riverside UC Riverside Electronic Theses and Dissertations Title Carboranes: Building Blocks for Materials and Ligand Development Permalink https://escholarship.org/uc/item/2vp9m2z6 Author Estrada, Jess Steven Publication Date 2017 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA RIVERSIDE Carboranes: Building Blocks for Materials and Ligand Development A Dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Chemistry by Jess Steven Estrada September 2017 Dissertation Committee: Dr. Vincent Lavallo, Chairperson Dr. Richard Hooley Dr. Pingyun Feng Copyright by Jess Steven Estrada 2017 The Dissertation Jess Steven Estrada is approved: Committee Chairperson University of California, Riverside Acknowledgements I would like to thank Dr. Vincent Lavallo for giving me the opportunity to join his lab and for all of his help and support throughout graduate school. I owe a lot of my success to you. I would also like to thank the amazing faculty at UCR for all of their help and knowledge they have provided. I truly enjoyed every class I took here at UCR. In addition to the faculty, the staff in the chemistry department has been amazing over the last five years and played a huge role in my success as well so I would specifically like to thank Dr. Borchardt, the NMR genius, Dr. Fook for all of his help with my great looking X-ray structures, Christina Youhas for answering literally every question I ever had and being so kind about answering. My lab mates, who created the most unique group of people probably in the history of UCR and that I’ve ever had the pleasure of working with. -

United States Patent Office Patented Jan
3,489,812 United States Patent Office Patented Jan. 13, 1970 2 3,489,812 and sufficient dialkyl sulfide to permit convenient stirring METHOD OF PREPARING BIS-(DIETHYL of the reactant mixture. The reaction should be carried SULFIDE)-DECABORANE out in the absence of air or other oxidizing gas, suitably Mervin D. Marshall, Fombell, and Richard M. Hunt and by using a vacuum or nonoxidizing cover gas such as Gerald T. Heiferan, Butler, Pa., assignors to Mine nitrogen or argon. Safety Appliances Company, a corporation of Pennsyl vania tion:The following examples are illustrative of this inven No Drawing. Filed Jan. 15, 1968, Ser. No. 697,596 Int, C. C07f 5/02 EXAMPLE I U.S. C. 260-606.5 1 Claim O A glass reactor equipped with a magnetic stirrer and connected to a glass vacuum apparatus was charged with ABSTRACT OF THE DISCLOSURE 3.513 g. of (NH4)2BioHo (22.6 millimols) and 40 ml. of diethyl sulfide, cooled to about 4° C. and evacuated. Bis-(diethyl sulfide)-decaborane is prepared by reac Measured aliquots of anhydrous hydrogen chloride (92. tion of ammonium decahydrodecarborate, HCl and di 15 immoles) were added in two equal increments. This reac ethyl sulfide. tion mixture was allowed to warm to room temperature and stirred for 72 hours. A white precipitate formed which Bis-(dialkyl sulfide)-decaboranes, such as bis-(dimethyl was filtered from the diethyl sulfide solution. The evapora sulfide)-decaborane, BioH12S (CH3)2)2, and bis-(diethyl tion of excess diethyl sulfide from the solution gave 4.596 sulfide)-decaborane, BioH12S (C2H5)2)2, have been used g. -

Nite States Patent O?Ice Patented Apr
3,030,423 nite States Patent O?ice Patented Apr. 17, 1962 1 2 cuprous chloride catalyst employed also can be varied 3,030,423 REACTION PRODUCTS 0F ACETYLENES widely, generally being in the range of from 0.005 to 0.5 AND DECABORANE mole of cuprous chloride per mole of decaborane and Starling K. Alley, Beverly Hills, Calif., and Otto Fuchs, preferably from 0.01 to 0.2 mole of cuprous chloride per Tonawanzia, N.Y., assignors to Olin Mathieson Chemi- 5 mole of decaborane. The reaction temperature can vary cal Corporation, a corporation of Virginia from 100 to 350° C. and preferably from 150 to 250° C. No Drawing. Filed Feb. 13, 1959, Ser. No. 793,211 The pressure can vary from atmospheric pressure to about 3 Claims. (Cl. 260-6065) 700 p.s.i.g., although closed system reactions are usually conducted at about 100 to 600 p.s.i.g. The reaction gen This invention relates to solid products obtained by 10 erally requires about 1 to 10 hours depending upon the the reaction of acetylene or methylacetylene with deca~ ratio of reactants and the temperature and pressure em borane. ployed. The solid products of this invention, when incorporated Although the reaction will proceed in the absence of a with suitable oxidizers such as ammonium perchlorate, solvent, a solvent common for the reactants but inert with potassium perchlorate, sodium perchlorate, etc., yield 15 respect to the reactants under the reaction conditions is solid propellants generally suitable for rocket power usually employed. Such solvents include aliphatic hydro plants and other jet propelled devices. -

Safety Data Sheet
SAFETY DATA SHEET Revision Date 14-February-2020 Revision Number 2 1. Identification Product Name Decaborane Cat No. : 87892 CAS-No 17702-41-9 Synonyms No information available Recommended Use Laboratory chemicals. Uses advised against Food, drug, pesticide or biocidal product use. Details of the supplier of the safety data sheet Company Importer/Distributor Manufacturer Fisher Scientific Alfa Aesar 112 Colonnade Road, Thermo Fisher Scientific Chemicals, Inc. Ottawa, ON K2E 7L6, 30 Bond Street, Ward Hill, MA 01835-8099 Canada Tel: 800-343-0660 Fax: 800-322-4757 Tel: 1-800-234-7437 Email: [email protected] www.alfa.com Emergency Telephone Number During normal business hours (Monday-Friday, 8am-7pm EST), call (800) 343-0660. After normal business hours, call Carechem 24 at (800) 579-7421. 2. Hazard(s) identification Classification WHMIS 2015 Classification Classified as hazardous under the Hazardous Products Regulations (SOR/2015-17) Flammable solids Category 2 Acute oral toxicity Category 3 Acute dermal toxicity Category 3 Acute Inhalation Toxicity Category 2 Physical Hazards Not Otherwise Classified Category 1 Risk of explosion if heated under confinement Label Elements Signal Word Danger Hazard Statements Flammable solid Fatal if inhaled Toxic if swallowed or in contact with skin ______________________________________________________________________________________________ Page 1 / 7 Decaborane Revision Date 14-February-2020 ______________________________________________________________________________________________ Risk of explosion if heated under confinement Precautionary Statements Manufacturer Alfa Aesar Thermo Fisher Scientific Chemicals, Inc. 30 Bond Street, Ward Hill, MA 01835-8099 Tel: 800-343-0660 Fax: 800-322-4757 Email: [email protected] www.alfa.com Prevention Keep away from heat, hot surfaces, sparks, open flames and other ignition sources. -

Tuning Photophysical Properties in Closo-Decaborane Cluster Derivatives
TUNING PHOTOPHYSICAL PROPERTIES IN CLOSO-DECABORANE CLUSTER DERIVATIVES by Mustapha B. Abdulmojeed A Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science in Chemistry Middle Tennessee State University 2020 Thesis Committee: Dr. Andrienne C. Friedli, Major Advisor Dr. Piotr Kaszynski, Committee Member Dr. Kevin Bicker, Committee Member Dr. Justin Miller, Committee Member ii Tuning Photophysical Properties in Closo-Decaborane Cluster Derivatives by Mustapha B. Abdulmojeed Approved: _____________________________________________ Dr. Andrienne C. Friedli, Committee Chair _____________________________________________ Dr. Piotr Kaszynski, Committee Member _____________________________________________ Dr. Kevin L. Bicker, Committee Member _____________________________________________ Dr. Justin M. Miller, Committee Member _____________________________________________ Dr. Paul G. Van Patten, Chair, Department of Chemistry _____________________________________________ Dr. David L. Butler, Dean, College of Graduate Studies ii iii ACKNOWLEDGEMENTS All thanks belong to God who has made it possible to complete this piece of work. Thanks to my wife for her love, care and understanding, taking care of our kids while I was away. Special thanks to my advisor, Dr. Andrienne Friedli for her support, guide and patiently delivered instructions through-out the course of this project. Dziękuję to my co- advisor, Dr. Piotr Kaszynski whose constant push and motivation from across the Atlantic Ocean kept me going, no amount of words could express my gratitude to both of you. Sincere appreciation also goes to Dr. Szymon Kapuscinski who is not just an excellent senior colleague but a great personality whose help in this work cannot be measured. I also want to thank MTSU chemistry department for the funding opportunity. I extend my gratitude to all faculty members in the department for creating a conducive and welcoming environment to learn and train as a chemist. -

And 1,7-Carborane Ligands for Boron Neutron Capture Therapy
YO t' Lg-3^o Ptatinum(Il) Complexes Containing lr2' and lr7-Carborane Ligands for Boron Neutron Capture Therapy A Thesis Submitted Towards the Degree of Doctor ofPhilosophy by Jean Ann Todd B.Sc. (Hons) * * a ADELAIDE * t UNIVERSITY t- AUSTRALIA cRucr L Department of Chemistry October 2001 Contents Acknowledgements IX Statement x Abstract xl Abbreviations xiii Chapter I : Introduction I l.l Cancer 1.1.1 Current Cancer Therapy 2 1.1.2 Binary Systems 2 l.l.2.l The Neutron Capture Reaction 2 1.2 Boron Neutron Capture Therapy J 1.2.1 The Advantages of BNCT J 1.2.2 Optimisation of BNCT 4 1.2.3 Application of BNCT 5 1.2.3.1 The Blood-Brain Barrier 5 1.2.4 Early Trials of BNCT 6 1.2.5 Recent Advances in BNCT 8 1.2.5.1The Carboranes 8 l.2.5.2Types of BNCT Agents 9 1.3 Tumour Selective Agents l0 1.3.1 Cellular Builcling Blocks l0 I 1.3.1. I Boron-Containing Nucleic Acid Precursors I .3.1.2Boron-Containing Amino Acids 1.3.1.3 Lipids, Phospholipids and Carbohydrates 1.3.2 Lipoproteins 1.3.3 Liposomes 1.3.4 Receptor / Antigen Binders 1.3.4.1Antibodies l-3.4.2Growth Factors 1.3.4.3 Hormones 1.3.5 Porphyrins 1.4 DNA Targeting Agents for BNCT 1.4.1 AlkylatingAgents L4.2 DNA Intercalators 1.4.3 Groove Binders I.4.4 Polyamines 1.4.5 Nucleic Acid Precursors 1.5 Background of platinum anti-cancer drugs 1.5.1 Cisplatin 1.5.1 .1 Activity and Action of Cisplatin 1.5.1.2 Problems Associated with the Use of Cisplatin in Cancer Therapy 1.5.2 New Generations of Platinum Anti-tumour Complexes 1.5.2.1Carboplatin 1.5.2.2 Other analogues of Cisplatin and Carboplatin- -
Chapter 3 Molecular Shape and Structure
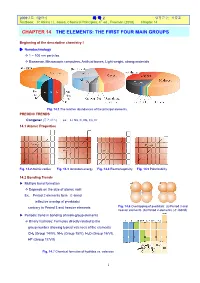
2009년도 제2학기 화 학 2 담당교수: 신국조 Textbook: P. Atkins / L. Jones, Chemical Principles, 4th ed., Freeman (2008) Chapter 14 CHAPTER 14 THE ELEMENTS: THE FIRST FOUR MAIN GROUPS Beginning of the descriptive chemistry ! ▶ Nanotechnology Æ 1 ~ 100 nm particles Æ Biosensor, Microscopic computers, Artificial bones, Light weight, strong materials Fig. 14.1 The relative abundances of the principal elements. PREODIC TRENDS Congener (동족원소) ex. Li, Na, K, Rb, Cs, Fr 14.1 Atomic Properties Fig. 14.2 Atomic radius Fig. 14.3 Ionization energy Fig. 14.4 Electronegativity Fig. 14.5 Polarizability 14.2 Bonding Trends ▶ Multiple bond formation Æ Depends on the size of atomic radii Ex. Period 2 elements form π -bond (effective overlap of p-orbitals) contrary to Period 3 and heavier elements Fig. 14.6 Overlapping of p-orbitals: (a) Period 3 and heavier elements. (b) Period 2 elements (π -bond) ▶ Periodic trend in bonding of main-group elements Æ Binary hydrides: Formulas directly related to the group number showing typical valences of the elements CH4 (Group 14/IV), NH3 (Group 15/V), H2O (Group 16/VI), HF (Group 17/VII) Fig. 14.7 Chemical formulas of hydrides vs. valences 1 2009년도 제2학기 화 학 2 담당교수: 신국조 Textbook: P. Atkins / L. Jones, Chemical Principles, 4th ed., Freeman (2008) Chapter 14 ▶ Binary hydrides Saline hydrides (이온성 수소화물): s-block elements, white, high m.p., crystals, portable fuel, H– ∆ 2 K(sg) +⎯H2 ( ) ⎯→2 KH(s) Metallic hydrides: d-block elements, black, conducting powder, portable fuel ∆ 2 Cu(sg) +⎯H2 ( ) ⎯→2 CuH(s) Molecular hydrides: non-metal elements, low melting volatile Brønsted acid NH3, HX, hydrocarbons Fig. -

Nomenclature of Organic Chemistry. IUPAC Recommendations and Preferred Names 2013
International Union of Pure and Applied Chemistry Division VIII Chemical Nomenclature and Structure Representation Division Nomenclature of Organic Chemistry. IUPAC Recommendations and Preferred Names 2013. Prepared for publication by Henri A. Favre and Warren H. Powell, Royal Society of Chemistry, ISBN 978-0-85404-182-4 Chapter P-6 APPLICATIONS TO SPECIFIC CLASSES OF COMPOUNDS (continued) (P-66 to P-69) (continued from P-60 to P-65) P-60 Introduction P-61 Substitutive nomenclature: prefix mode P-62 Amines and imines P-63 Hydroxy compounds, ethers, peroxols, peroxides and chalcogen analogues P-64 Ketones, pseudoketones and heterones, and chalcogen analogues P-65 Acids and derivatives P-66 Amides, hydrazides, nitriles, aldehydes P-67 Oxoacids used as parents for organic compounds P-68 Nomenclature of other classes of compounds P-69 Organometallic compounds P-66 AMIDES, IMIDES, HYDRAZIDES, NITRILES, AND ALDEHYDES, P-66.0 Introduction P-66.1 Amides P-66.2 Imides P-66.3 Hydrazides P-66.4 Amidines, amidrazones, hydrazidines, and amidoximes (amide oximes) P-66.5 Nitriles P-66.6 Aldehydes P-66.0 INTRODUCTION The classes dealt with in this Section have in common the fact that their retained names are derived from those of acids by changing the ‘ic acid’ ending to a class name, for example ‘amide’, ‘ohydrazide’, ‘nitrile’, or ‘aldehyde’. Their systematic names are formed substitutively by the suffix mode using one of two types of suffix, one that includes the carbon atom, for example, ‘carbonitrile’ for –CN, and one that does not, for example, ‘-nitrile’ for –(C)N. Amidines are named as amides, hydrazidines as hydrazides, and amidrazones as amides or hydrazides. -

United States Patent Office Patented Oct
3,152,867 United States Patent Office Patented Oct. 13, 1964 2 actor was connected to the high vacuum system. The 3,152,867 amine was cooled to -196° C. by placing a liquid nitro PREPARATEON OF DECABORANE gen bath: around the tube, and the reaction tube was George Nobit Tyson, Jr. Claremont, Calif., assignor to evacuated. Then 2.10 millimoles of pentaborane (9) Olin Mathieson Chemical Corporation, a corporation were condensed on top of the diphenylamine in the re of Virginia - . action tube. The reaction vessel was sealed off from the No Drawing. Fied Apr. 2, 1957, Ser. No. 650,282 high vacuum system by means of a stop cock and was al 3 Claims. (C. 23-294) lowed to warm to room temperature. As the diphenyl This invention relates to the preparation of decaborane amine and pentaborane (9) melted together, a liquid and provides a method for production of decaborane. O formed. ... - from the reaction of pentaborane (9) and amines. The reaction tube which was still sealed off from the Decaborane is a stable, white, crystalline material with Vacuum system was treated as follows: the bottom quar a melting point of 99.5 C. Its boiling point is 213 C. ter of the tube was placed into a hot oil bath and held at atmospheric pressure and its density is 0.94 gram per at a temperature of 80-100° C. for two hours. The cc. at 25 C. The vapor pressure of this compound is 5 liquid in the tube which was originally clear at room 19.0 mm.