Crosstalk Between Cannabinoid Receptors and Epidermal
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cannabinoid As Potential Aromatase Inhibitor Through Molecular Modeling and Screening for Anti-Cancer Activity
Cannabinoid as Potential Aromatase Inhibitor Through Molecular Modeling and Screening for Anti-Cancer Activity Sudipta Baroi1, Achintya Saha2, Ritesh Bachar3 and Sitesh C Bachar4 1Department of Pharmaceutical Technology, Faculty of Pharmacy, University of Dhaka Dhaka-1000, Bangladesh 2Department of Chemical Technology, Pharmaceutical & Fine Chemical Technology Division University of Calcutta, India 3Department of Pharmacy, School of Science and Engineering, University of Information Technology and Sciences (UITS), Dhaka-1212, Bangladesh 4Department of Pharmacy, Faculty of Pharmacy, University of Dhaka, Dhaka-1000, Bangladesh (Received: April 15, 2020; Accepted: June 2, 2020; Published (web): June 28, 2020) ABSTRACT: Inhibition of aromatase (CYTP450), a key enzyme in the estrogen biosynthesis, could result in regression of estrogen-dependent tumors and even prevent the promotion of breast cancer. The present research has been designed for searching a potent chemical moiety from natural sources to inhibit aromatase enzyme, the over- functionality of which causes the breast cancer. Cannabis sativa contains a very much promising group of cannabinoids with more than 66 compounds with reported anticancer property and for the search of a target specific potent aromatase inhibitor, 61 cannabinoids from C. sativa were selected. The Structures Data File (SDF) of these ligand molecules were subjected to docking studies at the binding site of aromatase X-ray crystallographic structure based on lower resolution of the protein crystal structure and higher docking accuracy, predicted by calculating the correlation between experimental activities and Glide dock scores and compared with the standard aromatase ligand androstenedione and aromatase inhibitor fadrozole with existing drug for breast cancer treatment. The best docked pose of each ligand was selected on the basis of the highest dock score related to the binding free energies of the internal dataset compounds as compared to their observed activities. -

CBD (Cannabidiol)
TRANSPORTATION RESEARCH BOARD Driving Toward the Truth - Dispelling the Myths About Cannabis Products February 10, 2021 @NASEMTRB #TRBwebinar PDH Certification The Transportation Research Board has met the standards and Information: requirements of the Registered Continuing Education Providers •1.5 Professional Development Program. Credit earned on completion Hour (PDH) – see follow-up of this program will be reported to email for instructions RCEP. A certificate of completion will •You must attend the entire be issued to participants that have registered and attended the entire webinar to be eligible to receive session. As such, it does not include PDH credits content that may be deemed or •Questions? Contact Reggie construed to be an approval or Gillum at [email protected] endorsement by RCEP. #TRBwebinar Learning Objectives 1. Identify impacts of the Farm Bill on use of THC and CBD products 2. Describe the toxicology of THC and CBD products 3. Discuss how THC and CBD products affect driving performance and crash risk #TRBwebinar TRB Standing Committee on Impairment in Transportation (ACS50) TRB Webinar: Driving Toward the Truth - Dispelling the Myths About Cannabis Products Dr. Barry K. Logan Executive Director, Center for Forensic Science Research and Education (CFSRE); Senior Vice President of Forensic Sciences, and Chief Scientist at NMS Labs Michelle Peace, Ph.D. Associate Professor and PI, Laboratory for Forensic Toxicology Research Department of Forensic Science, Virginia Commonwealth University Dr. Darrin Grondel Vice President, -

Medical Marijuana: High Expectations
Medical Marijuana: High Expectations Dr. Ally Dering-Anderson, RP University of Nebraska College of Pharmacy August 2019 What we know What we don’t Speaker Contact Information Dr. Ally Dering-Anderson, RP Clinical Associate Professor of Pharmacy University of Nebraska College of Pharmacy 986145 Medical Center College of Pharmacy Department of Pharmacy Practice Omaha, NE 68198-6145 [email protected] Speaker Disclosures Dr. Ally is on the faculty of the University of Nebraska College of Pharmacy Department of Pharmacy Practice She is the Senior Partner of BBfN, a health and safety consulting company She has no conflict in regard to medical use of cannabinoids “Off Label Use” In order to assure that there are no problems with the accreditation of this program nor problems caused for the accrediting body here goes: There are 3 approved products containing one or more chemicals found in the marijuana plant. Epidiolex (cannabidiol) Marinol Cesamet We will NOT discuss uses for these 3 approved products beyond the labeling. We will discuss cannabinoids not found in drug products currently approved by the FDA. The concept of being “off label” for these products is illogical, as there is no label to follow. Got it? Bibliography: #’s 1, 2, 3 A note from the CE clinical reviewers … “Medical marijuana is considered a controversial topic in CME. When a CME activity includes information about an approach to diagnosis or treatment that is not generally accepted, it is allowable to facilitate debate and discussion about the approach, but it is not allowable to advocate for the test or treatment, or teach clinicians how or when to use it.” Trust me, I am not advocating for any test - - in fact, I’m going to slam one. -

What Is Delta-8 THC?? Cannabinoid Chemistry 101
What is Delta-8 THC?? Cannabinoid Chemistry 101 National Conference on Weights and Measures Annual Meeting - Rochester, NY Matthew D. Curran, Ph.D. July 21, 2021 Disclaimer Just to be clear… • I am a chemist and not a lawyer so: • This presentation will not discuss the legal aspects of Δ8-THC or DEA’s current position. • This presentation will not discuss whether Δ8-THC is considered “synthetic” or “naturally occurring.” • This is not a position statement on any issues before the NCWM. • Lastly, this should only be considered a scientific sharing exercise. Florida Department of Agriculture and Consumer Services 2 Cannabis in Florida Cannabis Syllabus • What is Cannabis? • “Mother” Cannabinoid • Decarboxylation • Relationship between CBD and THC • What does “Total” mean? • Dry Weight vs. Wet Weight • What does “Delta-9” mean? • Relationship between “Delta-8” and “Delta-9” • CBD to Delta-8 THC • Cannabinoid Chemistry 202… Florida Department of Agriculture and Consumer Services 3 Cannabis Cannabis • Cannabis sativa is the taxonomic name for the plant. • The concentration of Total Δ9-Tetrahydrocannabinol (Total Δ9-THC) is critical when considering the varieties of Cannabis sativa. • Hemp – (Total Δ9-THC) 0.3% or less • Not really a controversial term, “hemp” • Marijuana/cannabis – (Total Δ9-THC) Greater than 0.3% • Controversial term, “marijuana” • Some states prohibit the use of this term whereas some states have it in their laws. • Some states use the term “cannabis.” • Not italicized • Lower case “c” Florida Department of Agriculture and -

2 Cannabis Sativa L
CANNABIS SATIVA 29 2 Cannabis sativa L. Common Names Almindelig hamp Denmark Harilik kanep Slovenia Asa Japan Haschischpflanze Germany Bang Egypt Hash United Kingdom Bhaang India Hashas Turkey Bhaango Nepal Hashish Morocco Canamo indico Spain Hemp United Kingdom Canapa indica Italy Hennep Netherlands Canhamo Portugal Hind kinnabi Turkey Cares Nepal Huo ma cao China Chanvre cultive France Huo ma China Chanvre de l’Inde France Indian hemp United Kingdom Chanvre France Indische hennep Netherlands Chanvrier sauvage France Indischer hanf Germany Charas India Indisk hamp Sweden Churras India Kannabis Finland Da ma cao China Kannabisu Japan Da ma ren China Kerp Albania Da ma China Kinnab Turkey Dagga South Africa Konopie siewne Poland Dansk pot Denmark Konopie Poland Echter hanf Germany Konoplja Slovenia Esrar Turkey Kultur hanf Germany Gaanjaa Nepal Maconha Portugal Gajiimaa Nepal Marihana Netherlands Ganja Guyana Marihouava Greece Ganja India Marihuana Poland Grifa Spain Marihuana Bulgaria Hachis Spain Marihuana Croatia Hamp Denmark Marihuana Czech Republic Hamp Norway Marihuana Denmark Hampa Sweden Marihuana France Hampjurt Iceland Marihuana Germany Hamppu Finland Marihuana Hungary Hanf Germany Marihuana Mexico From: Medicinal Plants of the World, vol. 3: Chemical Constituents, Traditional and Modern Medicinal Uses By: I. A. Ross © Humana Press Inc., Totowa, NJ 29 30 MEDICINAL PLANTS OF THE WORLD Marihuana Russia Porkanchaa Thailand Marihuana Serbia Pot Denmark Marihuana Spain Qinnib Arabic countries Marihuana Ukraine Riesen hanf Germany Marihuana United States Seruma erva Portugal Marijuana France Taima Japan Marijuana Italy Til Arabic countries Marijuana Mexico Vrai chanvre France Marijuana Portugal Weed Guyana Marijuana Sweden Xian ma China Mashinin Japan Ye ma China Navadna konoplja Slovenia BOTANICAL DESCRIPTION types of plants are numerous varieties that Cannabis sativa is an annual herb of the differ from the main one in height, extent MORACEAE family that grows to 5 m tall. -

Invited Review Article การใช้กัญชาเพื่อประโยชน์ทางการแพทย์
Medical Uses of Cannabis IJPS Sripanidkulchai B. Vol. 15 No. 4 October – December 2019 _______________________________________________________________________________________________ Invited Review Article การใช้กัญชาเพื่อประโยชน์ทางการแพทย์ 1 บงั อร ศรีพานิชกลุ ชยั * 1 ศูนย์วิจัยและพัฒนาผลิตภัณฑ์สุขภาพจากสมุนไพร คณะเภสัชศาสตร์ มหาวิทยาลัยขอนแก่น ต.ในเมือง อ.เมือง จ.ขอนแก่น 40002 * ติดต่อผนู้ ิพนธ ์ : ศูนย์วิจัยและพัฒนาผลิตภัณฑ์สุขภาพจากสมุนไพร คณะเภสัชศาสตร์ มหาวิทยาลัยขอนแก่น ต.ในเมือง อ.เมือง จ.ขอนแก่น 40002 อีเมล: [email protected] บทคดั ย่อ การใช้กัญชาเพื่อประโยชน์ทางการแพทย์ บังอร ศรีพานิชกุลชัย1* ว. เภสัชศาสตร์อีสาน 2562; 15(4) : 1-26 รับบทความ : 11 กันยายน 2562 แก้ไขบทความ: 21 ตุลาคม 2562 ตอบรับ: 14 พฤศจิกายน 2562 กัญชาเป็นพืชในวงศ์แคนนาบาซีที่ขึ้นได้ดีในเขตอากาศร้อน เช่น ประเทศไทย และจัดเป็นพืชเสพติดที่เคยใช้เป็นยารักษาโรค แต่ต่อมามกี ารหา้ มใชเ้ น่ืองจากทา ใหเ้ กดิ การเสพตดิ การศกึ ษาต่อมาพบว่าสารสา คญั กลุ่มแคนนาบนิ อยดม์ ฤี ทธติ์ ่อร่างกายหลายประการ โดยที่สารสาคัญที่อยู่ในความสนใจคือ ทีเอชซี (delta-9-tetracannabinol, THC) ซง่ึ มฤี ทธติ์ ่อจติ ประสาทและซบี ดี ี (cannabidiol, CBD) ที่ ไม่มฤี ทธติ์ ่อจติ ประสาท ปัจจุบนั มผี ลงานวจิ ยั ช้แี นะว่าสารทงั้ สองชนิดสามารถน ามาใช้เป็นยารกั ษาโรคและอาการแสดงไดห้ ลายชนิด ได้แก่ อาการปวดประสาท อาการหดเกร็งของกล้ามเนื้อ ต้านการชัก อาการคลื่นไส้อาเจียน เพิ่มความอยากอาหาร โดยมีกลไกการทางาน ผ่านตัวรับเอนโดแคนตาบินอยด์ที่มีบทบาทสาคัญในการควบคุมภาวะโฮมิโอสิสของร่างกาย บทความนี้จึงมีวัตถุประสงค์ที่จะรวบรวมสรุป องค์ความรู้ที่เป็นปัจจุบันเกี่ยวกับกัญชาเพื่อให้เกิดความเข้าใจที่ถูกต้องในด้านการน ากัญชามาใช้เป็นประโยชน์ทางการแพทย์ -
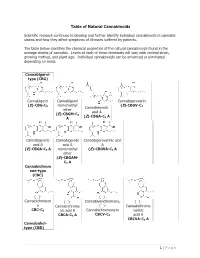
Table of Natural Cannabinoids
Table of Natural Cannabinoids Scientific research continues to develop and further identify individual cannabinoids in cannabis strains and how they affect symptoms of illnesses suffered by patients. The table below identifies the chemical properties of the natural cannabinoids found in the average strains of cannabis. Levels of each of these chemicals will vary with varietal strain, growing method, and plant age. Individual cannabinoids can be enhanced or eliminated depending on need. Cannabigerol- type (CBG) Cannabigerol Cannabigerol Cannabigerovarin (E)-CBG-C monomethyl (E)-CBGV-C 5 Cannabinerolic 3 ether acid A (E)-CBGM-C 5 (Z)-CBGA-C A A 5 Cannabigerolic Cannabigerolic Cannabigerovarinic acid acid A acid A A (E)-CBGA-C5 A monomethyl (E)-CBGVA-C3 A ether (E)-CBGAM- C5 A Cannabichrom ene-type (CBC) (±)- (±)- Cannabichromen (±)- Cannabivarichromene, (±)- e Cannabichrome (±)- Cannabichrome CBC-C5 nic acid A Cannabichromevarin varinic CBCA-C5 A CBCV-C3 acid A CBCVA-C3 A Cannabidiol- type (CBD) 1 | Page (−)-Cannabidiol Cannabidiol Cannabidiol-C4 (−)- Cannabidiorc CBD-C5 momomethyl CBD-C4 Cannabidivarin ol ether CBDV-C3 CBD-C1 CBDM-C5 Cannabidiolic Cannabidivarini acid c acid CBDA-C5 CBDVA-C3 Cannabinodiol- type (CBND) Cannabinodiol Cannabinodivar CBND-C5 in CBND-C3 Tetrahydrocan nabinol-type (THC) 9 9 9 Δ - Δ - Δ - Δ9- Tetrahydrocanna Tetrahydrocan Tetrahydrocannabivarin 9 Tetrahydrocan binol nabinol-C4 Δ -THCV-C3 9 9 nabiorcol Δ -THC-C5 Δ -THC-C4 9 Δ -THCO-C1 9 9 Δ -Tetrahydro- Δ9-Tetrahydro- Δ -Tetrahydro- Δ9-Tetrahydro- cannabinolic -

Dr Ben Jansen General Practitioner Sports Doctor Bundall Medical Centre Gold Coast
Dr Ben Jansen General Practitioner Sports Doctor Bundall Medical Centre Gold Coast 16:30 - 17:25 WS #169: Medicinal Use of Cannabis 17:35 - 18:30 WS #181: Medicinal Use of Cannabis (Repeated) Medicinal Cannabis 101 Dr Ben Jansen Director @ Burleigh Heads Cannabis FRNZCGP FRACGP FRCUCP Medicinal Cannabis 101 Medicinal Cannabis 101 Medicinal Cannabis 101 Take Home Messages Medicinal Cannabis does not need to get the patient “high” / neuropsychologically altered Start low and titrate dose to effect: THC 1mg No direct mortality from cannabis use CBD, among other cannabinoids, modulate THC effects The non-psychoactive natural raw acid forms of the cannabinoids are being used for CB2 reception modulation and preventing oxidative stress (among other effects) Public Opinion On Regulation? Prohibition Medication Restricted Medication Open Recreational Use* (still requires a level of quality and control for sale) PubMed search 2017/4/1 31945 results for Cannabis, or Marijuana, or THC, or CBD Compared to 23924 for Paracetamol, or Acetaminophen Compared to 7511 for Metoprolol Compared to 2598 for Ramipril Mortality 69000+ Opioid deaths estimated Worldwide in 2014 by the WHO. 22598 deaths from Opioid Pain Relievers in the USA 2015. National Center on Health Statistics, CDC WONDER 56,000 emergency room visits and 26,000 hospitalizations yearly from Paracetamol, and 458 deaths in the USA. Nourjah P et al. Pharmacoepidemiol Drug Saf. 2006 Jun;15(6):398-405. 3,200 deaths annually as a result of NSAID-induced GI bleeding in the USA. Tarone RE et al. Am J Ther. 2004;11(1):17-25. Zero deaths from Cannabis, ever. -

WO 2018/071581 Al 19 April 2018 (19.04.2018) W !P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2018/071581 Al 19 April 2018 (19.04.2018) W !P O PCT (51) International Patent Classification: A61K 9/16 (2006.01) A61K 31/352 (2006.01) A61K 9/48 (2006.01) (21) International Application Number: PCT/US20 17/056201 (22) International Filing Date: 11 October 2017 ( 11.10.2017) (25) Filing Language: English (26) Publication Language: English (30) Priority Data: 62/406,980 12 October 2016 (12.10.2016) US (71) Applicant: COLUMBIA CARE, LLC [US/US]; 745 Fifth Avenue, Suite 1701, New York, NY 10151 (US). (72) Inventor: DELY, Aaron; 10655 NW 29th Terrace, Miami, FL 33 172 (US). (74) Agent: ACETO, Joseph, F.; Attorney at Law, 1617 Newark Road, Kennett Square, PA 19348 (US). (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Regulate Cannabinoid Products. (Public)
GENERAL ASSEMBLY OF NORTH CAROLINA SESSION 2021 H 1 HOUSE BILL 818 Short Title: Regulate Cannabinoid Products. (Public) Sponsors: Representatives Sasser, Humphrey, and McNeely (Primary Sponsors). For a complete list of sponsors, refer to the North Carolina General Assembly web site. Referred to: Agriculture, if favorable, Rules, Calendar, and Operations of the House May 5, 2021 1 A BILL TO BE ENTITLED 2 AN ACT TO DIRECT THE DEPARTMENT OF AGRICULTURE AND CONSUMER 3 SERVICES TO ESTABLISH A VOLUNTARY LICENSING PROGRAM FOR 4 CANNABINOID-RELATED COMPOUNDS. 5 The General Assembly of North Carolina enacts: 6 SECTION 1. G.S. 106-121 reads as rewritten: 7 "§ 106-121. Definitions and general consideration. 8 For the purpose of this Article: 9 (1) The term "advertisement" means all representations disseminated in any 10 manner or by any means, other than by labeling, for the purposes of inducing, 11 or which are likely to induce, directly or indirectly, the purchase of food, 12 drugs, devices or cosmetics. 13 (1a) The term "cannabinoid-related compounds" means any phytocannabinoid 14 found in hemp, including, but not limited to, tetrahydrocannabinol (THC), 15 tetrahydrocannabinolic acid (THCA), cannabidiol (CBD), cannabidiolic acid 16 (CBDA), cannabinol (CBN), cannabigerol (CBG), cannabichromene (CBC), 17 cannabicyclol (CBL), cannabivarin (CBV), tetrahydrocannabivarin (THCV), 18 cannabidivarin (CBDV), cannabichromevarin (CBCV), cannabigerovarin 19 (CBGV), cannabigerol monomethyl ether (CBGM), cannabielsoin (CBE), or 20 cannabicitran (CBT). Cannabinoids do not include synthetic cannabinoids. 21 (1a)(1b) The term "color" includes black, white, and intermediate grays. 22 (1b)(1c) The term "color additive" means a material which: 23 …." 24 SECTION 2. G.S. -

Pre-Harvest Sampling Protocol Publication Date: August 25, 2020
1 Hemp Pre-Harvest Sampling Protocol Publication Date: August 25, 2020 Hemp or hemp is the Cannabis sativa L. and any part of that plant, including the seeds thereof and all derivatives, extracts, cannabinoids, isomers, acids, salts, and salts of isomers, whether growing or not, with a delta-9 tetrahydrocannabinol (THC) concentration of no more than 0.3 percent on a dry weight basis. In Vermont, hemp is considered an "agricultural product" when grown by an individual that is registered with Vermont Agency of Agriculture, Food & Markets (VAAFM) as part of its Hemp Program. VAAFM through its Hemp Program, authorized under 6 VSA, Chapter 34, registers hemp growers and processors. The VAAFM requires registrants to maintain sampling and testing records to indicate proof of compliance for potency and contaminants. A grower registrant must maintain records for a period of three years for all harvest lots grown in Vermont, and a processor registrant must also maintain records of pre-harvest sampling and potency and contaminants testing for harvest lots that come into their possession. To be sufficient to meet the requirements for potency and contaminant sampling and testing under the Vermont Hemp Program Rules (VHPR) sampling and testing must be conducted as described in this protocol. Section 1 Definitions: 1.1. Acceptable potency level means a hemp crop that has a delta-9 tetrahydrocannabinol concentration of 0.3 percent or less on a dry weight basis. This initial requirement accords with the federal 2014 Farm Bill. As an additional policy limitation implemented to protect public safety, the Agency also requires that the total theoretical tetrahydrocannabinol concentration not exceed one percent on a dry weight basis. -

International Journal of Pharmacology Research
Vol 2|Issue 2| 2012 | 88-108. ISSN 2249 - 7641 Print ISSN 2249 - 765X International Journal of Pharmacology Research www.ijprjournal.org AUTOPHARMACOLOGY- A REVIEW Saravana Kumar A*, Gandhimathi R *Department of Pharmacology, Sri Venkateswara College of Pharmacy, RVS Nagar, Chittoor, Andhra Pradesh, India. ABSTRACT Autopharmacology relates to the scientific study of the regulation of body functions by the activity of its naturally existent (or endogenous) chemical factors of the tissues. A more restricted definition would consider substances that were first identified as external agents which had a documented action on physiological functions, but later were discovered as existing as endogenous factors. The best example is the class of endorphins, which, as it name implies, were discovered to exist in the brain and have specific receptors in it, by investigations on the mechanism of action of opioids, such as morphine. A research area where autopharmacology principles assumed great importance was that of pain and inflammation, due to the great number of endogenous messengers, transmitters and modulators involved in their complex response at molecular and cellular level. Therefore the present review focused on endogenous substances that could fall under the concept of autopharmacology. Keywords: Autopharmacology, Endogenous substances, Body functions. INTRODUCTION Angiotensin Historically, the first approach to the concept of Secretin autopharmacology began with British physiologist and Gastrin pharmacologist Henry Dale in the 1910s, discovered the Cholecystokinin role of acetylcholine in synaptic transmission, and later Histamine proved by Austrian physiologist Otto Loewi, to be the Cannabinoids neurotransmitter involved in the proximal synapses of the Substance P autonomic nervous system (initially named Vagusstoff by The main scientific criterion for an Loewi, and later identified as acetylcholine).