RNA-Seq Analysis of Broiler Liver Transcriptome Reveals Novel Responses to High Ambient Temperature Derrick Jamaal Coble Iowa State University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Towards Mutation-Specific Precision Medicine in Atypical Clinical
International Journal of Molecular Sciences Review Towards Mutation-Specific Precision Medicine in Atypical Clinical Phenotypes of Inherited Arrhythmia Syndromes Tadashi Nakajima * , Shuntaro Tamura, Masahiko Kurabayashi and Yoshiaki Kaneko Department of Cardiovascular Medicine, Gunma University Graduate School of Medicine, Maebashi 371-8511, Gunma, Japan; [email protected] (S.T.); [email protected] (M.K.); [email protected] (Y.K.) * Correspondence: [email protected]; Tel.: +81-27-220-8145; Fax: +81-27-220-8158 Abstract: Most causal genes for inherited arrhythmia syndromes (IASs) encode cardiac ion channel- related proteins. Genotype-phenotype studies and functional analyses of mutant genes, using heterol- ogous expression systems and animal models, have revealed the pathophysiology of IASs and enabled, in part, the establishment of causal gene-specific precision medicine. Additionally, the utilization of induced pluripotent stem cell (iPSC) technology have provided further insights into the patho- physiology of IASs and novel promising therapeutic strategies, especially in long QT syndrome. It is now known that there are atypical clinical phenotypes of IASs associated with specific mutations that have unique electrophysiological properties, which raises a possibility of mutation-specific precision medicine. In particular, patients with Brugada syndrome harboring an SCN5A R1632C mutation exhibit exercise-induced cardiac events, which may be caused by a marked activity-dependent loss of R1632C-Nav1.5 availability due to a marked delay of recovery from inactivation. This suggests that the use of isoproterenol should be avoided. Conversely, the efficacy of β-blocker needs to be examined. Patients harboring a KCND3 V392I mutation exhibit both cardiac (early repolarization syndrome and Citation: Nakajima, T.; Tamura, S.; paroxysmal atrial fibrillation) and cerebral (epilepsy) phenotypes, which may be associated with a Kurabayashi, M.; Kaneko, Y. -
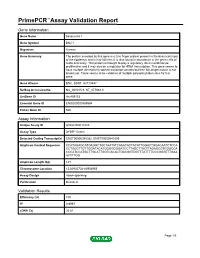
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name basonuclin 1 Gene Symbol BNC1 Organism Human Gene Summary The protein encoded by this gene is a zinc finger protein present in the basal cell layer of the epidermis and in hair follicles. It is also found in abundance in the germ cells of testis and ovary. This protein is thought to play a regulatory role in keratinocyte proliferation and it may also be a regulator for rRNA transcription. This gene seems to have multiple alternatively spliced transcript variants but their full-length nature is not known yet. There seems to be evidence of multiple polyadenylation sites for this gene. Gene Aliases BNC, BSN1, HsT19447 RefSeq Accession No. NC_000015.9, NT_077661.3 UniGene ID Hs.459153 Ensembl Gene ID ENSG00000169594 Entrez Gene ID 646 Assay Information Unique Assay ID qHsaCID0017223 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000345382, ENST00000541809 Amplicon Context Sequence CCATAGAGCATGAGGCTGCTAATATCAAACACTACATTGGACTGGACAATCTCCA CCTGGCTTGTTGGATACATGGGGGGGATCCTTAGCTTACTTAGAGCGTGGGCCA CCCATCCATGCTTGCATTGGTCACACTGACGGTGGTTTATTTTCCCGGGTTTGAA ACTTTGG Amplicon Length (bp) 141 Chromosome Location 15:83935724-83936955 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9997 cDNA Cq 26.81 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 84.5 gDNA Cq 39.42 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report BNC1, Human Amplification -

Investigating Unexplained Deaths for Molecular Autopsies
The author(s) shown below used Federal funding provided by the U.S. Department of Justice to prepare the following resource: Document Title: Investigating Unexplained Deaths for Molecular Autopsies Author(s): Yingying Tang, M.D., Ph.D, DABMG Document Number: 255135 Date Received: August 2020 Award Number: 2011-DN-BX-K535 This resource has not been published by the U.S. Department of Justice. This resource is being made publically available through the Office of Justice Programs’ National Criminal Justice Reference Service. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Final Technical Report NIJ FY 11 Basic Science Research to Support Forensic Science 2011-DN-BX-K535 Investigating Unexplained Deaths through Molecular Autopsies May 28, 2017 Yingying Tang, MD, PhD, DABMG Principal Investigator Director, Molecular Genetics Laboratory Office of Chief Medical Examiner 421 East 26th Street New York, NY, 10016 Tel: 212-323-1340 Fax: 212-323-1540 Email: [email protected] Page 1 of 41 This resource was prepared by the author(s) using Federal funds provided by the U.S. Department of Justice. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Abstract Sudden Unexplained Death (SUD) is natural death in a previously healthy individual whose cause remains undetermined after scene investigation, complete autopsy, and medical record review. SUD affects children and adults, devastating families, challenging medical examiners, and is a focus of research for cardiologists, neurologists, clinical geneticists, and scientists. -

S41467-020-18249-3.Pdf
ARTICLE https://doi.org/10.1038/s41467-020-18249-3 OPEN Pharmacologically reversible zonation-dependent endothelial cell transcriptomic changes with neurodegenerative disease associations in the aged brain Lei Zhao1,2,17, Zhongqi Li 1,2,17, Joaquim S. L. Vong2,3,17, Xinyi Chen1,2, Hei-Ming Lai1,2,4,5,6, Leo Y. C. Yan1,2, Junzhe Huang1,2, Samuel K. H. Sy1,2,7, Xiaoyu Tian 8, Yu Huang 8, Ho Yin Edwin Chan5,9, Hon-Cheong So6,8, ✉ ✉ Wai-Lung Ng 10, Yamei Tang11, Wei-Jye Lin12,13, Vincent C. T. Mok1,5,6,14,15 &HoKo 1,2,4,5,6,8,14,16 1234567890():,; The molecular signatures of cells in the brain have been revealed in unprecedented detail, yet the ageing-associated genome-wide expression changes that may contribute to neurovas- cular dysfunction in neurodegenerative diseases remain elusive. Here, we report zonation- dependent transcriptomic changes in aged mouse brain endothelial cells (ECs), which pro- minently implicate altered immune/cytokine signaling in ECs of all vascular segments, and functional changes impacting the blood–brain barrier (BBB) and glucose/energy metabolism especially in capillary ECs (capECs). An overrepresentation of Alzheimer disease (AD) GWAS genes is evident among the human orthologs of the differentially expressed genes of aged capECs, while comparative analysis revealed a subset of concordantly downregulated, functionally important genes in human AD brains. Treatment with exenatide, a glucagon-like peptide-1 receptor agonist, strongly reverses aged mouse brain EC transcriptomic changes and BBB leakage, with associated attenuation of microglial priming. We thus revealed tran- scriptomic alterations underlying brain EC ageing that are complex yet pharmacologically reversible. -

The DNA Methylation Landscape of Glioblastoma Disease Progression Shows Extensive Heterogeneity in Time and Space
bioRxiv preprint doi: https://doi.org/10.1101/173864; this version posted August 9, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space Johanna Klughammer1*, Barbara Kiesel2,3*, Thomas Roetzer3,4, Nikolaus Fortelny1, Amelie Kuchler1, Nathan C. Sheffield5, Paul Datlinger1, Nadine Peter3,4, Karl-Heinz Nenning6, Julia Furtner3,7, Martha Nowosielski8,9, Marco Augustin10, Mario Mischkulnig2,3, Thomas Ströbel3,4, Patrizia Moser11, Christian F. Freyschlag12, Jo- hannes Kerschbaumer12, Claudius Thomé12, Astrid E. Grams13, Günther Stockhammer8, Melitta Kitzwoegerer14, Stefan Oberndorfer15, Franz Marhold16, Serge Weis17, Johannes Trenkler18, Johanna Buchroithner19, Josef Pichler20, Johannes Haybaeck21,22, Stefanie Krassnig21, Kariem Madhy Ali23, Gord von Campe23, Franz Payer24, Camillo Sherif25, Julius Preiser26, Thomas Hauser27, Peter A. Winkler27, Waltraud Kleindienst28, Franz Würtz29, Tanisa Brandner-Kokalj29, Martin Stultschnig30, Stefan Schweiger31, Karin Dieckmann3,32, Matthias Preusser3,33, Georg Langs6, Bernhard Baumann10, Engelbert Knosp2,3, Georg Widhalm2,3, Christine Marosi3,33, Johannes A. Hainfellner3,4, Adelheid Woehrer3,4#§, Christoph Bock1,34,35# 1 CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria. 2 Department of Neurosurgery, Medical University of Vienna, Vienna, Austria. 3 Comprehensive Cancer Center, Central Nervous System Tumor Unit, Medical University of Vienna, Austria. 4 Institute of Neurology, Medical University of Vienna, Vienna, Austria. 5 Center for Public Health Genomics, University of Virginia, Charlottesville VA, USA. 6 Department of Biomedical Imaging and Image-guided Therapy, Computational Imaging Research Lab, Medical University of Vi- enna, Vienna, Austria. -

Identification of Novel DNA-Methylated Genes
Prostate Cancer and Prostatic Disease (2013) 16, 292–300 & 2013 Macmillan Publishers Limited All rights reserved 1365-7852/13 www.nature.com/pcan ORIGINAL ARTICLE Identification of novel DNA-methylated genes that correlate with human prostate cancer and high-grade prostatic intraepithelial neoplasia JM Devaney1, S Wang2,3, S Funda1, J Long2, DJ Taghipour2, R Tbaishat3, P Furbert-Harris2,4, M Ittmann5 and B Kwabi-Addo2,3 BACKGROUND: Prostate cancer (PCa) harbors a myriad of genomic and epigenetic defects. Cytosine methylation of CpG-rich promoter DNA is an important mechanism of epigenetic gene inactivation in PCa. There is considerable amount of data to suggest that DNA methylation-based biomarkers may be useful for the early detection and diagnosis of PCa. In addition, candidate gene- based studies have shown an association between specific gene methylation and alterations and clinicopathologic indicators of poor prognosis in PCa. METHODS: To more comprehensively identify DNA methylation alterations in PCa initiation and progression, we examined the methylation status of 485 577 CpG sites from regions with a broad spectrum of CpG densities, interrogating both gene-associated and non-associated regions using the recently developed Illumina 450K methylation platform. RESULTS: In all, we selected 33 promoter-associated novel CpG sites that were differentially methylated in high-grade prostatic intraepithelial neoplasia and PCa in comparison with benign prostate tissue samples (false discovery rate-adjusted P-value o0.05; b-value X0.2; fold change 41.5). Of the 33 genes, hierarchical clustering analysis demonstrated BNC1, FZD1, RPL39L, SYN2, LMX1B, CXXC5, ZNF783 and CYB5R2 as top candidate novel genes that are frequently methylated and whose methylation was associated with inactivation of gene expression in PCa cell lines. -

Mapping Protein Interactions of Sodium Channel Nav1.7 Using 2 Epitope-Tagged Gene Targeted Mice
bioRxiv preprint doi: https://doi.org/10.1101/118497; this version posted March 20, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Mapping protein interactions of sodium channel NaV1.7 using 2 epitope-tagged gene targeted mice 3 Alexandros H. Kanellopoulos1†, Jennifer Koenig1, Honglei Huang2, Martina Pyrski3, 4 Queensta Millet1, Stephane Lolignier1, Toru Morohashi1, Samuel J. Gossage1, Maude 5 Jay1, John Linley1, Georgios Baskozos4, Benedikt Kessler2, James J. Cox1, Frank 6 Zufall3, John N. Wood1* and Jing Zhao1†** 7 1Molecular Nociception Group, WIBR, University College London, Gower Street, 8 London WC1E 6BT, UK. 9 2Mass Spectrometry Laboratory, Target Discovery Institute, University of Oxford, The 10 Old Road Campus, Oxford, OX3 7FZ, UK. 11 3Center for Integrative Physiology and Molecular Medicine, Saarland University, 12 Kirrbergerstrasse, Bldg. 48, 66421 Homburg, Germany. 13 4Division of Bioscience, University College London, Gower Street, London WC1E 6BT, UK.14 15 †These authors contributed equally to directing this work. 16 *Correspondence author. Tel: +44 207 6796 954; E-mail: [email protected] 17 **Corresponding author: Tel: +44 207 6790 959; E-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/118497; this version posted March 20, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 18 Abstract 19 The voltage-gated sodium channel NaV1.7 plays a critical role in pain pathways. 20 Besides action potential propagation, NaV1.7 regulates neurotransmitter release, 21 integrates depolarizing inputs over long periods and regulates transcription. -

Atrial Fibrillation (ATRIA) Study
European Journal of Human Genetics (2014) 22, 297–306 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg REVIEW Atrial fibrillation: the role of common and rare genetic variants Morten S Olesen*,1,2,4, Morten W Nielsen1,2,4, Stig Haunsø1,2,3 and Jesper H Svendsen1,2,3 Atrial fibrillation (AF) is the most common cardiac arrhythmia affecting 1–2% of the general population. A number of studies have demonstrated that AF, and in particular lone AF, has a substantial genetic component. Monogenic mutations in lone and familial AF, although rare, have been recognized for many years. Presently, mutations in 25 genes have been associated with AF. However, the complexity of monogenic AF is illustrated by the recent finding that both gain- and loss-of-function mutations in the same gene can cause AF. Genome-wide association studies (GWAS) have indicated that common single-nucleotide polymorphisms (SNPs) have a role in the development of AF. Following the first GWAS discovering the association between PITX2 and AF, several new GWAS reports have identified SNPs associated with susceptibility of AF. To date, nine SNPs have been associated with AF. The exact biological pathways involving these SNPs and the development of AF are now starting to be elucidated. Since the first GWAS, the number of papers concerning the genetic basis of AF has increased drastically and the majority of these papers are for the first time included in a review. In this review, we discuss the genetic basis of AF and the role of both common and rare genetic variants in the susceptibility of developing AF. -

Gene-Based and Pathway-Based Genome-Wide Association Study of Alcohol Dependence
• 111 • Shanghai Archives of Psychiatry, 2015, Vol. 27, No. 2 •Original research article• Gene-based and pathway-based genome-wide association study of alcohol dependence Lingjun ZUO1, Clarence K. ZHANG2,3*, Frederick G. SAYWARD4,5, Kei-Hoi CHEUNG4, Kesheng WANG6, John H. KRYSTAL1, Hongyu ZHAO2, Xingguang LUO1* Background: The organization of risk genes within signaling pathways may provide clues about the converging neurobiological effects of risk genes for alcohol dependence. Aims: Identify risk genes and risk gene pathways for alcohol dependence. Methods: We conducted a pathway-based genome-wide association study (GWAS) of alcohol dependence using a gene-set-rich analytic approach. Approximately one million genetic markers were tested in the discovery sample which included 1409 European-American (EA) alcohol dependent individuals and 1518 EA healthy comparison subjects. An additional 681 African-American (AA) cases and 508 AA healthy subjects served as the replication sample. Results: We identified several genome-wide replicable risk genes and risk pathways that were significantly associated with alcohol dependence. After applying the Bonferroni correction for multiple testing, the ‘cell- extracellular matrix interactions’ pathway (p<2.0E-4 in EAs) and the PXN gene (which encodes paxillin) (p=3.9E-7 in EAs) within this pathway were the most promising risk factors for alcohol dependence. There were also two nominally replicable pathways enriched in alcohol dependence-related genes in both EAs (0.015≤p≤0.035) and AAs (0.025≤p≤0.050): the ‘Na+/Cl- dependent neurotransmitter transporters’ pathway and the ‘other glycan degradation’ pathway. Conclusions: These findings provide new evidence highlighting several genes and biological signaling processes that may be related to the risk for alcohol dependence. -

Spatial Distribution of Leading Pacemaker Sites in the Normal, Intact Rat Sinoa
Supplementary Material Supplementary Figure 1: Spatial distribution of leading pacemaker sites in the normal, intact rat sinoatrial 5 nodes (SAN) plotted along a normalized y-axis between the superior vena cava (SVC) and inferior vena 6 cava (IVC) and a scaled x-axis in millimeters (n = 8). Colors correspond to treatment condition (black: 7 baseline, blue: 100 µM Acetylcholine (ACh), red: 500 nM Isoproterenol (ISO)). 1 Supplementary Figure 2: Spatial distribution of leading pacemaker sites before and after surgical 3 separation of the rat SAN (n = 5). Top: Intact SAN preparations with leading pacemaker sites plotted during 4 baseline conditions. Bottom: Surgically cut SAN preparations with leading pacemaker sites plotted during 5 baseline conditions (black) and exposure to pharmacological stimulation (blue: 100 µM ACh, red: 500 nM 6 ISO). 2 a &DUGLDFIoQChDQQHOV .FQM FOXVWHU &DFQDG &DFQDK *MD &DFQJ .FQLS .FQG .FQK .FQM &DFQDF &DFQE .FQM í $WSD .FQD .FQM í .FQN &DVT 5\U .FQM &DFQJ &DFQDG ,WSU 6FQD &DFQDG .FQQ &DFQDJ &DFQDG .FQD .FQT 6FQD 3OQ 6FQD +FQ *MD ,WSU 6FQE +FQ *MG .FQN .FQQ .FQN .FQD .FQE .FQQ +FQ &DFQDD &DFQE &DOP .FQM .FQD .FQN .FQG .FQN &DOP 6FQD .FQD 6FQE 6FQD 6FQD ,WSU +FQ 6FQD 5\U 6FQD 6FQE 6FQD .FQQ .FQH 6FQD &DFQE 6FQE .FQM FOXVWHU V6$1 L6$1 5$ /$ 3 b &DUGLDFReFHSWRUV $GUDF FOXVWHU $GUDD &DY &KUQE &KUP &KJD 0\O 3GHG &KUQD $GUE $GUDG &KUQE 5JV í 9LS $GUDE 7SP í 5JV 7QQF 3GHE 0\K $GUE *QDL $QN $GUDD $QN $QN &KUP $GUDE $NDS $WSE 5DPS &KUP 0\O &KUQD 6UF &KUQH $GUE &KUQD FOXVWHU V6$1 L6$1 5$ /$ 4 c 1HXURQDOPURWHLQV -
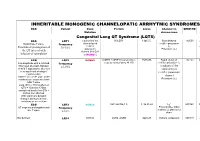
Inheritable Arrhytmia Syndromes
INHERITABLE MONOGENIC CHANNELOPATIC ARRHYTMIC SYNDROMES ECG Variant Gene Protein Locus Channel on OMIM NO IP Mutation chromosome Congenital Long QT Syndrome (LQTS) ECG LQT1 α subunit of the KvLQT1 11p15.5. Slow delayed 192500 AD slow delayed rectifier potassium or Broad base T wave. Frequency rectifier I AR Paradoxical prolongation of 30-35% ks potassium Potassium (IKs) the QT interval with channel (KvLQT1 infusion of epinephrine or KCNQ1) ECG LQT2 KCNH2 (HERG + MiRP1) human ether- 7q35q36 Rapid delayed 152.427 AD a-go-go related gene HERG rectifier potassium I Low-amplitude with a notched, Frequency kr bifurcated alternant, biphasic 25-30% α subunit of the or bifid T appearance due to a rapid delayed very significant slowing of rectifier potassium repolarization. channel KCNH2 on L413P and L559H mutations are associated with Potassium (IKr) bifid T wave Long. QTc > 470ms affected QTc = 450ms to 470ms considered border line QTc < 450ms non affected. With significant dynamic changes during heart rate variations or on exertion + ECG LQT3 SCN5A hH1 and NaV1.5 3, 3p 21-24 INa 600163 AD Prolonged I + influx ST segment prolongation and Frequency Na late T wave. in phase 2, plateau or 5-10% dome Not defined LQT4 KCNJ2 ANK2, ANKB 4q25-27 Sodium, potassium 600919 AR and calcium + Not defined LQT5 KCNE1 minK K Efflux 176261 AD Not defined LQT6 KCNE2 MiRP1 603796 MiRP1 Frequency rare Modest prolongation of QT LQT7 Andersen KCNJ2 Kir2.1 Ik1 Reduction 170390 AD interval, prominent U wave, Great reduction in I Tawil-syndrome k1 frequent PVCs, PVT, result in the bidirectional VT. -

The Limb-Girdle Muscular Dystrophies and the Dystrophinopathies Review Article
Review Article 04/25/2018 on mAXWo3ZnzwrcFjDdvMDuzVysskaX4mZb8eYMgWVSPGPJOZ9l+mqFwgfuplwVY+jMyQlPQmIFeWtrhxj7jpeO+505hdQh14PDzV4LwkY42MCrzQCKIlw0d1O4YvrWMUvvHuYO4RRbviuuWR5DqyTbTk/icsrdbT0HfRYk7+ZAGvALtKGnuDXDohHaxFFu/7KNo26hIfzU/+BCy16w7w1bDw== by https://journals.lww.com/continuum from Downloaded Downloaded from Address correspondence to https://journals.lww.com/continuum Dr Stanley Jones P. Iyadurai, Ohio State University, Wexner The Limb-Girdle Medical Center, Department of Neurology, 395 W 12th Ave, Columbus, OH 43210, Muscular Dystrophies and [email protected]. Relationship Disclosure: by mAXWo3ZnzwrcFjDdvMDuzVysskaX4mZb8eYMgWVSPGPJOZ9l+mqFwgfuplwVY+jMyQlPQmIFeWtrhxj7jpeO+505hdQh14PDzV4LwkY42MCrzQCKIlw0d1O4YvrWMUvvHuYO4RRbviuuWR5DqyTbTk/icsrdbT0HfRYk7+ZAGvALtKGnuDXDohHaxFFu/7KNo26hIfzU/+BCy16w7w1bDw== Dr Iyadurai has received the Dystrophinopathies personal compensation for serving on the advisory boards of Allergan; Alnylam Stanley Jones P. Iyadurai, MSc, PhD, MD; John T. Kissel, MD, FAAN Pharmaceuticals, Inc; CSL Behring; and Pfizer, Inc. Dr Kissel has received personal ABSTRACT compensation for serving on a consulting board of AveXis, Purpose of Review: The classic approach to identifying and accurately diagnosing limb- Inc; as journal editor of Muscle girdle muscular dystrophies (LGMDs) relied heavily on phenotypic characterization and & Nerve; and as a consultant ancillary studies including muscle biopsy. Because of rapid advances in genetic sequencing for Novartis AG. Dr Kissel has received research/grant methodologies,