Mapping Protein Interactions of Sodium Channel Nav1.7 Using 2 Epitope-Tagged Gene Targeted Mice
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Analysis of the Shaker Gene Complex of Drosophila Melanogaster
Copyright 0 1990 by the Genetics Society of America Genetic Analysisof the Shaker Gene Complexof Drosophila melanogaster A. Ferriis,* S. LLamazares,* J. L. de la Pornpa,* M. A. Tanouye? and0. Pongs* *Institute Cajal, CSIC, 28006 Madrid, Spain, +California Instituteof Technology, Pasadena, California 91 125, and 'Ruhr Universitat,Bochum, Federal Republic of Germany Manuscript received May 10, 1989 Accepted for publication March 3, 1990 ABSTRACT The Shaker complex (ShC) spans over 350 kb in the 16F region of the X chromosome. It can be dissected by means of aneuploids into three main sections: the maternal effect (ME), the viable (V) and the haplolethal (HL) regions. The mutational analysis of ShC shows a high density of antimorphic mutations among 12 lethal complementation groupsin addition to 14 viable alleles. The complex is the structural locus of a family of potassium channels as well as a number of functions relevant to the biology of the nervous system. The constituents of ShC seem to be linked by functional relationships in view of the similarity of the phenotypes, antimorphic nature of their mutations and the behavior in transheterozygotes. We discuss the relationship between the genetic organization of ShC and the functional coupling of potassium currents with the other functions encodedin the complex. HAKER was the first behavioral mutant detected do not appear tohave the capability of generating, by S in Drosophila melanogaster (CATSCH1944). It was themselves, gated ionic channels. named after another mutantwith a similar phenotype K+ currents are known to be themost diverse class isolated earlier in Drosophila funebris (LUERS 1936). of ionic currents in terms of kinetics, pharmacology The original phenotype was described as the trem- and sensitivity (HILLE 1984; RUDY 1988).Also, these bling of appendagesin the anesthetized fly. -

Targeted Genes and Methodology Details for Neuromuscular Genetic Panels
Targeted Genes and Methodology Details for Neuromuscular Genetic Panels Reference transcripts based on build GRCh37 (hg19) interrogated by Neuromuscular Genetic Panels Next-generation sequencing (NGS) and/or Sanger sequencing is performed Motor Neuron Disease Panel to test for the presence of a mutation in these genes. Gene GenBank Accession Number Regions of homology, high GC-rich content, and repetitive sequences may ALS2 NM_020919 not provide accurate sequence. Therefore, all reported alterations detected ANG NM_001145 by NGS are confirmed by an independent reference method based on laboratory developed criteria. However, this does not rule out the possibility CHMP2B NM_014043 of a false-negative result in these regions. ERBB4 NM_005235 Sanger sequencing is used to confirm alterations detected by NGS when FIG4 NM_014845 appropriate.(Unpublished Mayo method) FUS NM_004960 HNRNPA1 NM_031157 OPTN NM_021980 PFN1 NM_005022 SETX NM_015046 SIGMAR1 NM_005866 SOD1 NM_000454 SQSTM1 NM_003900 TARDBP NM_007375 UBQLN2 NM_013444 VAPB NM_004738 VCP NM_007126 ©2018 Mayo Foundation for Medical Education and Research Page 1 of 14 MC4091-83rev1018 Muscular Dystrophy Panel Muscular Dystrophy Panel Gene GenBank Accession Number Gene GenBank Accession Number ACTA1 NM_001100 LMNA NM_170707 ANO5 NM_213599 LPIN1 NM_145693 B3GALNT2 NM_152490 MATR3 NM_199189 B4GAT1 NM_006876 MYH2 NM_017534 BAG3 NM_004281 MYH7 NM_000257 BIN1 NM_139343 MYOT NM_006790 BVES NM_007073 NEB NM_004543 CAPN3 NM_000070 PLEC NM_000445 CAV3 NM_033337 POMGNT1 NM_017739 CAVIN1 NM_012232 POMGNT2 -

Towards Mutation-Specific Precision Medicine in Atypical Clinical
International Journal of Molecular Sciences Review Towards Mutation-Specific Precision Medicine in Atypical Clinical Phenotypes of Inherited Arrhythmia Syndromes Tadashi Nakajima * , Shuntaro Tamura, Masahiko Kurabayashi and Yoshiaki Kaneko Department of Cardiovascular Medicine, Gunma University Graduate School of Medicine, Maebashi 371-8511, Gunma, Japan; [email protected] (S.T.); [email protected] (M.K.); [email protected] (Y.K.) * Correspondence: [email protected]; Tel.: +81-27-220-8145; Fax: +81-27-220-8158 Abstract: Most causal genes for inherited arrhythmia syndromes (IASs) encode cardiac ion channel- related proteins. Genotype-phenotype studies and functional analyses of mutant genes, using heterol- ogous expression systems and animal models, have revealed the pathophysiology of IASs and enabled, in part, the establishment of causal gene-specific precision medicine. Additionally, the utilization of induced pluripotent stem cell (iPSC) technology have provided further insights into the patho- physiology of IASs and novel promising therapeutic strategies, especially in long QT syndrome. It is now known that there are atypical clinical phenotypes of IASs associated with specific mutations that have unique electrophysiological properties, which raises a possibility of mutation-specific precision medicine. In particular, patients with Brugada syndrome harboring an SCN5A R1632C mutation exhibit exercise-induced cardiac events, which may be caused by a marked activity-dependent loss of R1632C-Nav1.5 availability due to a marked delay of recovery from inactivation. This suggests that the use of isoproterenol should be avoided. Conversely, the efficacy of β-blocker needs to be examined. Patients harboring a KCND3 V392I mutation exhibit both cardiac (early repolarization syndrome and Citation: Nakajima, T.; Tamura, S.; paroxysmal atrial fibrillation) and cerebral (epilepsy) phenotypes, which may be associated with a Kurabayashi, M.; Kaneko, Y. -

Expression Profiling of Ion Channel Genes Predicts Clinical Outcome in Breast Cancer
UCSF UC San Francisco Previously Published Works Title Expression profiling of ion channel genes predicts clinical outcome in breast cancer Permalink https://escholarship.org/uc/item/1zq9j4nw Journal Molecular Cancer, 12(1) ISSN 1476-4598 Authors Ko, Jae-Hong Ko, Eun A Gu, Wanjun et al. Publication Date 2013-09-22 DOI http://dx.doi.org/10.1186/1476-4598-12-106 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Ko et al. Molecular Cancer 2013, 12:106 http://www.molecular-cancer.com/content/12/1/106 RESEARCH Open Access Expression profiling of ion channel genes predicts clinical outcome in breast cancer Jae-Hong Ko1, Eun A Ko2, Wanjun Gu3, Inja Lim1, Hyoweon Bang1* and Tong Zhou4,5* Abstract Background: Ion channels play a critical role in a wide variety of biological processes, including the development of human cancer. However, the overall impact of ion channels on tumorigenicity in breast cancer remains controversial. Methods: We conduct microarray meta-analysis on 280 ion channel genes. We identify candidate ion channels that are implicated in breast cancer based on gene expression profiling. We test the relationship between the expression of ion channel genes and p53 mutation status, ER status, and histological tumor grade in the discovery cohort. A molecular signature consisting of ion channel genes (IC30) is identified by Spearman’s rank correlation test conducted between tumor grade and gene expression. A risk scoring system is developed based on IC30. We test the prognostic power of IC30 in the discovery and seven validation cohorts by both Cox proportional hazard regression and log-rank test. -

Investigating Unexplained Deaths for Molecular Autopsies
The author(s) shown below used Federal funding provided by the U.S. Department of Justice to prepare the following resource: Document Title: Investigating Unexplained Deaths for Molecular Autopsies Author(s): Yingying Tang, M.D., Ph.D, DABMG Document Number: 255135 Date Received: August 2020 Award Number: 2011-DN-BX-K535 This resource has not been published by the U.S. Department of Justice. This resource is being made publically available through the Office of Justice Programs’ National Criminal Justice Reference Service. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Final Technical Report NIJ FY 11 Basic Science Research to Support Forensic Science 2011-DN-BX-K535 Investigating Unexplained Deaths through Molecular Autopsies May 28, 2017 Yingying Tang, MD, PhD, DABMG Principal Investigator Director, Molecular Genetics Laboratory Office of Chief Medical Examiner 421 East 26th Street New York, NY, 10016 Tel: 212-323-1340 Fax: 212-323-1540 Email: [email protected] Page 1 of 41 This resource was prepared by the author(s) using Federal funds provided by the U.S. Department of Justice. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Abstract Sudden Unexplained Death (SUD) is natural death in a previously healthy individual whose cause remains undetermined after scene investigation, complete autopsy, and medical record review. SUD affects children and adults, devastating families, challenging medical examiners, and is a focus of research for cardiologists, neurologists, clinical geneticists, and scientists. -

Allosteric Features of KCNQ1 Gating Revealed by Alanine Scanning Mutagenesis
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biophysical Journal Volume 100 February 2011 885–894 885 Allosteric Features of KCNQ1 Gating Revealed by Alanine Scanning Mutagenesis Li-Juan Ma, Iris Ohmert, and Vitya Vardanyan* Institut fu¨r Neurale Signalverarbeitung, Zentrum fu¨r Molekulare Neurobiologie, Universita¨t Hamburg, Hamburg, Germany ABSTRACT Controlled opening and closing of an ion-selective pathway in response to changes of membrane potential is a fundamental feature of voltage-gated ion channels. In recent decades, various details of this process have been revealed with unprecedented precision based on studies of prototypic potassium channels. Though current scientific efforts are focused more on a thorough description of voltage-sensor movement, much less is known about the similarities and differences of the gating mechanisms among potassium channels. Here, we describe the peculiarities of the KCNQ1 gating process in parallel comparison to Shaker. We applied alanine scanning mutagenesis to the S4-S5 linker and pore region and followed the regular- ities of gating perturbations in KCNQ1. We found a fractional constitutive conductance for wild-type KCNQ1. This component increased significantly in mutants with considerably leftward-shifted steady-state activation curves. In contrast to Shaker,no correlation between V1/2 and Z parameters was observed for the voltage-dependent fraction of KCNQ1. Our experimental find- ings are explained by a simple allosteric gating scheme with voltage-driven and voltage-independent transitions. Allosteric features are discussed in the context of extreme gating adaptability of KCNQ1 upon interaction with KCNE b-subunits. -

Atrial Fibrillation (ATRIA) Study
European Journal of Human Genetics (2014) 22, 297–306 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg REVIEW Atrial fibrillation: the role of common and rare genetic variants Morten S Olesen*,1,2,4, Morten W Nielsen1,2,4, Stig Haunsø1,2,3 and Jesper H Svendsen1,2,3 Atrial fibrillation (AF) is the most common cardiac arrhythmia affecting 1–2% of the general population. A number of studies have demonstrated that AF, and in particular lone AF, has a substantial genetic component. Monogenic mutations in lone and familial AF, although rare, have been recognized for many years. Presently, mutations in 25 genes have been associated with AF. However, the complexity of monogenic AF is illustrated by the recent finding that both gain- and loss-of-function mutations in the same gene can cause AF. Genome-wide association studies (GWAS) have indicated that common single-nucleotide polymorphisms (SNPs) have a role in the development of AF. Following the first GWAS discovering the association between PITX2 and AF, several new GWAS reports have identified SNPs associated with susceptibility of AF. To date, nine SNPs have been associated with AF. The exact biological pathways involving these SNPs and the development of AF are now starting to be elucidated. Since the first GWAS, the number of papers concerning the genetic basis of AF has increased drastically and the majority of these papers are for the first time included in a review. In this review, we discuss the genetic basis of AF and the role of both common and rare genetic variants in the susceptibility of developing AF. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Engineering Biosynthetic Excitable Tissues from Unexcitable Cells for Electrophysiological and Cell Therapy Studies
ARTICLE Received 11 Nov 2010 | Accepted 5 Apr 2011 | Published 10 May 2011 DOI: 10.1038/ncomms1302 Engineering biosynthetic excitable tissues from unexcitable cells for electrophysiological and cell therapy studies Robert D. Kirkton1 & Nenad Bursac1 Patch-clamp recordings in single-cell expression systems have been traditionally used to study the function of ion channels. However, this experimental setting does not enable assessment of tissue-level function such as action potential (AP) conduction. Here we introduce a biosynthetic system that permits studies of both channel activity in single cells and electrical conduction in multicellular networks. We convert unexcitable somatic cells into an autonomous source of electrically excitable and conducting cells by stably expressing only three membrane channels. The specific roles that these expressed channels have on AP shape and conduction are revealed by different pharmacological and pacing protocols. Furthermore, we demonstrate that biosynthetic excitable cells and tissues can repair large conduction defects within primary 2- and 3-dimensional cardiac cell cultures. This approach enables novel studies of ion channel function in a reproducible tissue-level setting and may stimulate the development of new cell-based therapies for excitable tissue repair. 1 Department of Biomedical Engineering, Duke University, Durham, North Carolina 27708, USA. Correspondence and requests for materials should be addressed to N.B. (email: [email protected]). NatURE COMMUNicatiONS | 2:300 | DOI: 10.1038/ncomms1302 | www.nature.com/naturecommunications © 2011 Macmillan Publishers Limited. All rights reserved. ARTICLE NatUre cOMMUNicatiONS | DOI: 10.1038/ncomms1302 ll cells express ion channels in their membranes, but cells a b with a significantly polarized membrane that can undergo e 0 a transient all-or-none membrane depolarization (action A 1 potential, AP) are classified as ‘excitable cells’ . -

Familial Hyperkalemic Hypertension
Disease of the Month Familial Hyperkalemic Hypertension Juliette Hadchouel,* Ce´line Delaloy,* Se´bastien Faure´,† Jean-Michel Achard,† and Xavier Jeunemaitre* *Colle`ge de France, Paris; INSERM U36, Paris; AP-HP, Department of Genetics, Hoˆpital Europe´en Georges Pompidou; University Paris-Descartes, Faculty of Medicine, Paris, France; and †Division of Nephrology and Department of Physiology, Limoges University Hospital, Limoges, France J Am Soc Nephrol 17: 208–217, 2006. doi: 10.1681/ASN.2005030314 amilial hyperkalemic hypertension (FHHt) syndrome biochemical abnormalities and age or BP, which seemed to (1,2), also known as Gordon syndrome (3) or pseudohy- depend primarily on age in affected individuals (11). This F poaldosteronism type 2 (4), is a rare inherited form of phenotypic variability, associated with sensitivity to thia- low-renin hypertension associated with hyperkalemia and hy- zides—which are widely used in hypertension—and with a low perchloremic metabolic acidosis in patients with a normal GFR probability of this rare disease’s being recognized by most (OMIM no. 145260). This monogenic form of arterial hyperten- doctors may have led to an underestimation of its frequency. sion has excited new interest since the discovery of a new, The mode of inheritance of the disease is consistent with unsuspected molecular pathway that is responsible for both the autosomal dominant transmission in most, if not all, of the biochemical abnormalities and the increase in BP observed. pedigrees reported. However, we have identified two families Genetic analysis has led to the identification of mutations in in which the parents of the affected individuals were first two genes that belong to a new family of kinases, the WNK cousins, suggesting possible autosomal recessive transmission. -

Spatial Distribution of Leading Pacemaker Sites in the Normal, Intact Rat Sinoa
Supplementary Material Supplementary Figure 1: Spatial distribution of leading pacemaker sites in the normal, intact rat sinoatrial 5 nodes (SAN) plotted along a normalized y-axis between the superior vena cava (SVC) and inferior vena 6 cava (IVC) and a scaled x-axis in millimeters (n = 8). Colors correspond to treatment condition (black: 7 baseline, blue: 100 µM Acetylcholine (ACh), red: 500 nM Isoproterenol (ISO)). 1 Supplementary Figure 2: Spatial distribution of leading pacemaker sites before and after surgical 3 separation of the rat SAN (n = 5). Top: Intact SAN preparations with leading pacemaker sites plotted during 4 baseline conditions. Bottom: Surgically cut SAN preparations with leading pacemaker sites plotted during 5 baseline conditions (black) and exposure to pharmacological stimulation (blue: 100 µM ACh, red: 500 nM 6 ISO). 2 a &DUGLDFIoQChDQQHOV .FQM FOXVWHU &DFQDG &DFQDK *MD &DFQJ .FQLS .FQG .FQK .FQM &DFQDF &DFQE .FQM í $WSD .FQD .FQM í .FQN &DVT 5\U .FQM &DFQJ &DFQDG ,WSU 6FQD &DFQDG .FQQ &DFQDJ &DFQDG .FQD .FQT 6FQD 3OQ 6FQD +FQ *MD ,WSU 6FQE +FQ *MG .FQN .FQQ .FQN .FQD .FQE .FQQ +FQ &DFQDD &DFQE &DOP .FQM .FQD .FQN .FQG .FQN &DOP 6FQD .FQD 6FQE 6FQD 6FQD ,WSU +FQ 6FQD 5\U 6FQD 6FQE 6FQD .FQQ .FQH 6FQD &DFQE 6FQE .FQM FOXVWHU V6$1 L6$1 5$ /$ 3 b &DUGLDFReFHSWRUV $GUDF FOXVWHU $GUDD &DY &KUQE &KUP &KJD 0\O 3GHG &KUQD $GUE $GUDG &KUQE 5JV í 9LS $GUDE 7SP í 5JV 7QQF 3GHE 0\K $GUE *QDL $QN $GUDD $QN $QN &KUP $GUDE $NDS $WSE 5DPS &KUP 0\O &KUQD 6UF &KUQH $GUE &KUQD FOXVWHU V6$1 L6$1 5$ /$ 4 c 1HXURQDOPURWHLQV -
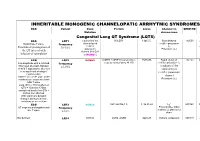
Inheritable Arrhytmia Syndromes
INHERITABLE MONOGENIC CHANNELOPATIC ARRHYTMIC SYNDROMES ECG Variant Gene Protein Locus Channel on OMIM NO IP Mutation chromosome Congenital Long QT Syndrome (LQTS) ECG LQT1 α subunit of the KvLQT1 11p15.5. Slow delayed 192500 AD slow delayed rectifier potassium or Broad base T wave. Frequency rectifier I AR Paradoxical prolongation of 30-35% ks potassium Potassium (IKs) the QT interval with channel (KvLQT1 infusion of epinephrine or KCNQ1) ECG LQT2 KCNH2 (HERG + MiRP1) human ether- 7q35q36 Rapid delayed 152.427 AD a-go-go related gene HERG rectifier potassium I Low-amplitude with a notched, Frequency kr bifurcated alternant, biphasic 25-30% α subunit of the or bifid T appearance due to a rapid delayed very significant slowing of rectifier potassium repolarization. channel KCNH2 on L413P and L559H mutations are associated with Potassium (IKr) bifid T wave Long. QTc > 470ms affected QTc = 450ms to 470ms considered border line QTc < 450ms non affected. With significant dynamic changes during heart rate variations or on exertion + ECG LQT3 SCN5A hH1 and NaV1.5 3, 3p 21-24 INa 600163 AD Prolonged I + influx ST segment prolongation and Frequency Na late T wave. in phase 2, plateau or 5-10% dome Not defined LQT4 KCNJ2 ANK2, ANKB 4q25-27 Sodium, potassium 600919 AR and calcium + Not defined LQT5 KCNE1 minK K Efflux 176261 AD Not defined LQT6 KCNE2 MiRP1 603796 MiRP1 Frequency rare Modest prolongation of QT LQT7 Andersen KCNJ2 Kir2.1 Ik1 Reduction 170390 AD interval, prominent U wave, Great reduction in I Tawil-syndrome k1 frequent PVCs, PVT, result in the bidirectional VT.