Cyanide Regeneration from Thiocyanate with the Use of Anion Exchange Resins
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Identification of Olefins As Thiocyanates
THE IDENTIFICATION OF OLEFI NS AS THIOCYANATES 1 .. .SEP 2"/ i 938 THE IDENTIFICATION OF OLEFINS AS THIOCYANAT ES/ ( ' By George A. Dysinger I\ Bachelor of Science Oklahoma Agricultural and Mechanical College Stillwater, Oklahoma 1937 Submitted to the Department of Chemistry Oklahoma Agricultural and Mechanical College In partial fulfillme.nt of the requirements for the Degree of MASTER OF SCIENCE 1938 . ... .. '.'' .. ~ . ..... .. • • • • • • • • J • : ... ·:· .· ~- . .. ,. r f • • • - • • • • •• J • •• ; • • • ii !)fp 27 1938 APPROVED: HeadOttinm~- of : e Department of Onemistry ~~~e~ 108550 iii ACKNOWLEDGEMENT The author wishes to expres.s his sincere appreciation to Dr. o. c. Dermer under whose direction and with whose help this work has been done. He also wishes to acknowledge the assistance rendered by Everett L. Ada.ms in supplying apparatus and chemicals. iv TABLE OF CONTENTS Page Introduotion • . .. • . • • • 1 Historical Review. • . • • . • . 2 Experimental . • • • • . • . • . • • 6 Discussion of Results . .. ao Summary • • • • . • • • 22 Bibliography • • • • • • • • • • • • • • • • • • 23 Autobiography • • • • • . • • • • • • • • • . • 24 1 INTRODUCTION At present, the identification of low boiling unsatu rated hydrocarbons which yield only liquid addition products with the halogens and hydrogen halides is often a matter of considerable difficulty. This f'aot is sufficient reason for the study here described. In some cases, espeoially among terpen.es, the compounds formed by adding NOCl, 1203 , or »2o4 at one or more of the double bonds have been used as derivatives but these are i nconvenient to make,. of un certain composition, and decidedly unstable! This work con- '\, sists of (l) a'ttempts to find a convenient samll scale meth- od for adding (S0N)2 to olef1ns, (2) the determinations of the melting points of the derivatives, and (3) quantitatiye analysis of derivatives to prove their structure and purity. -

Extraction of Cyanides from Waste Solutions of Cyanidation of .Lotation
Chemistry for Sustainable Development 12 (2004) 431436 431 Extraction of Cyanides from Waste Solutions of Cyanidation of lotation Concentrates from Kholbinskoye Gold Deposit A. A. KOCHANOV1, A. A. RYAZANTSEV1, A. A. BATOEVA2, D. B. ZHALSANOVA2, A. M. BADALYAN3 and O. V. POLYAKOV3 1Siberian Transport University, Ul. D. Kovalchuk 191, Novosibirsk 630049 (Russia) E-mail: [email protected] 2Baikal Institute of Nature Management, Siberian Branch of the Russian Academy of Sciences, Ul. Sakhyanovoy 6, Ulan Ude 670047 (Russia) 3A. V. Nikolaev Institute of Inorganic Chemistry, Siberian Branch of the Russian Academy of Sciences, Pr. Akademika Lavrentyeva 3, Novosibirsk 630090 (Russia) (Received April 21, 2003; in revised form January 6, 2004) Abstract Processes that occur during extraction of cyanides from cyanidation solutions using centrifugal bubbling apparatuses (CBA) as reactors have been studied. In the eddy chamber of CBA (ðÍ < 3), one can observe virtually complete removal of HCN from solution and precipitation of heavy metals in the form of insoluble x - 1 compounds. The electronic absorption spectra of solutions treated in CBA suggest that destruction of [Cu(CN)x] , + 2+ 2 oxidation of Cu to Cu by air oxygen, and oxidation of thiocyanates in the presence of SO23, forming 2 HCN and SO4 , are also accompanied by the emergence of stable intermediate products (SCN)2 and (SCN)x of oxidation in solution. INTRODUCTION formed after acidification of the waste process- ing solutions of cyanidation to ðÍ 62.5 fol- Cyanides are widely used in extraction of lowed by absorption of HCN by alkaline solu- precious metals, mainly fine gold and silver, tions. -

United States Patent Office Patented Feb
3,644,463 United States Patent Office Patented Feb. 22, 1972 2 agent that is introduced. Thus, for example, good yields 3,644,463 of 1,2-vinylene-bisthiocyanate are obtained by adding PRODUCTION OF ALEPHATIC 1,2-BISTHIOCYANATES acetylene gas, while the addition of ethylene results in the Richard Parke Welcher, Old Greenwich, Conn., assignor production of 1,2-dithiocyanoethane. The corresponding to American Cyanamid Company, Stamford, Conn. monoalkyl-substituted vinylene bisthiocyanates are pro No Drawing. Filed May 15, 1968, Ser. No. 729,375 duced when monoalkyl acetylene containing an alkyl radi Int, C. C07c 161/02 cal of from 1 to 16 carbn atoms are used, such as methyl U.S. C. 260-454 5 Claims acetylene, heptyne, octyne, octadecyne, and the like. Monoalkyl-dithiocyanoethanes are produced in similar O manner when monoalkylethylenes containing alkyl radi cals of 1-16 carbon atoms are used; typical of these are ABSTRACT OF THE DISCLOSURE propylene, isobutylene, octylethylene, hexadecyl-ethylene Aliphatic 1,2-bisthiocyanates are produced by first pre and the like. Dialkyl ethylenes may likewise be used, the paring a solution of thiocyanogen in a water-insoluble preferred reagents being those wherein the two alkyl sub liquid organic solvent such as toluene having an aqueous stituents taken together have a total of from 2 to 16 car solution of an inorganic halide admixed therewith, draw bon atoms. Aryl-substituted olefins such as styrene may ing off the aqueous phase, adding an alpha-olefin or acetyl also be used. The principles of the invention can also ene and reacting at a temperature below about 20° C. -

Thesis Reference
Thesis Experimental and theoretical studies of boron and hydrogen containing compounds in relation to potential hydrogen storage and ionic conduction applications SHARMA, Manish Abstract This thesis deals with the fundamental studies of some materials containing boron-hydrogen bonds which can potentially be used either as the hydrogen storage materials (M(BH4)2, M=Alkaline earth metal), as the solid electrolytes for batteries (Na2B12H12) or as reducing agents for CO2 (Mg(BH4)2). First part of thesis deals with borohydrides (BH4-). Synthesis and characterization of halide-free Sr(BH4)2, Ba(BH4)2 and Eu(BH4)2 is reported. Crystallographic study of these compounds helped in identifying several new phases and a new species metal borohydride hydride (M2(BH4)H3). In depth study of B-H bond breaking is reported via isotope exchange reaction in Ca(BH4)2.A practical example of borohydride as reducing agent is reported by showing the reduction of CO2 with gamma-Mg(BH4)2. The second part of the thesis focuses on closoboranes derived from the B12H122- ion. Compounds of this family have recently attracted great interest as solid ionic conductors for Li and Na ions.Results of DFT calculations on isolated B12H122- anions and halogen (F, Cl or Br) substituted anions were analysed in detail. Synthesis of Na2B12(SCN)H11 is [...] Reference SHARMA, Manish. Experimental and theoretical studies of boron and hydrogen containing compounds in relation to potential hydrogen storage and ionic conduction applications. Thèse de doctorat : Univ. Genève, 2017, no. Sc. 5101 DOI : 10.13097/archive-ouverte/unige:96376 URN : urn:nbn:ch:unige-963769 Available at: http://archive-ouverte.unige.ch/unige:96376 Disclaimer: layout of this document may differ from the published version. -

WSU Tri-Cities Laboratory Safety Manual
WSU Tri-Cities Laboratory Safety Manual December 2014 Contents Section I - Introduction ................................................................................................................... 1 A. Purpose ............................................................................................................................. 1 B. Scope and Application ..................................................................................................... 1 C. Implementation of the Chemical Hygiene Plan (CHP) .................................................... 2 D. Responsibilities ................................................................................................................ 3 Section II – General Policies and Recommendations ..................................................................... 5 A. Basic Rules and Procedures ............................................................................................. 5 B. Chemical Procurement, Storage, and Distribution .......................................................... 6 C. Exposure Monitoring ....................................................................................................... 8 D. Housekeeping ................................................................................................................. 10 E. Medical Surveillance ..................................................................................................... 10 F. Personal Protective Equipment ..................................................................................... -
UG Syllabus.Pdf
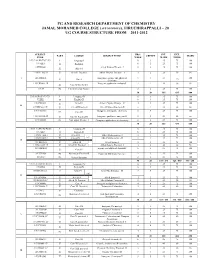
PG AND RESEARCH DEPARTMENT OF CHEMISTRY JAMAL MOHAMED COLLEGE (AUTONOMOUS), TIRUCHIRAPPALLI – 20 UG COURSE STRUCTURE FROM 2011-2012 SUBJECT HRS / INT. EXT. ` PART COURSE SUBJECT TITLE CREDIT MARK CODE WEEK MARK MARK 11U1LA1/H1/T1/F1/U1 I Language-I 6 3 25 75 100 11UILE1 6 3 25 75 100 II English-I 11UPH1301 Allied Physics Theory - I 5 3 25 75 100 III Allied-I 11UPH1302:1P III Allied-I-Practical Allied Physics Practical – I 3 2 20 30 50 11UCH1401 Inorganic, organic and physical 5 5 25 100 I III Core-I 75 chemistry 11UCH1402:1P Inorganic qualitative analysis-I 3 2 20 30 50 III Core-II-Practical-I 11U19 IV Environmental Studies 2 2 25 75 100 30 20 165 435 600 U2LA2/H2/T2/F2/U2 I Language-II 6 3 25 75 100 U2LE2 English-II 6 3 25 75 100 II 11UPH2303 III Allied-II Allied Physics Theory – II 4 3 25 75 100 II 11UPH2302:2P III Allied-II Practical Allied Physics Practical-II 3 2 20 30 50 11UCH2403 Inorganic and organic chemistry 6 5 25 75 100 III Core-III 11UCH2402:2P III Core-II Practical-II Inorganic qualitative analysis-II 3 2 20 30 50 11UCH2601 IV Non-major Elective-I Computer applications in chemistry 2 2 25 75 100 30 20 165 435 600 11U3LA3/H3/T3/F3/U3 I Language-III 6 3 25 75 100 11U3LE3 II English-III 6 3 25 75 100 11UMA3304:2 III Allied-III Allied Mathematics - I 5 3 25 75 100 (or) 11UMA3305:2 III Allied-1V Allied Mathematics - II 3 2 25 75 100 III 11UBO3304 III Allied-III Allied Botany-I 5 3 25 75 100 11UBO3305:1P III Allied-III Practical Allied Botany Practical-I 3 2 20 30 50 11UCH3404 organic and physical chemistry 6 5 25 75 100 III Core-IV -

Krok 2. Pharmacy
Sample test questions Krok 2 Pharmacy () Фармацевтична хiмiя 2 1. An analyst of a chemical laboratory A. Ammonium oxalate had received a glucose substance for B. Magnesium sulfate analysis. To check its quality he used a C. Potassium chloride polarimeter. The analyst measured: D. Sodium nitrite E. Ammonium molybdate A. Angle of rotation B. Refractive index 7. Optical rotation angle of substances is C. Optical density determined under the temperature of 20oC, D. Melting point in 1-decimeter-thick layer, with wavelength E. Specificgravity of sodium spectrum D-lines (λ = 589.3 nm), and is calculated with reference to a solution 2. The analytical laboratory received containing 1 g of the substance per ml. It is calcium chloride. What volumetric called: solution should be used for quantitative determination of this medicinal substance? A. Specific optical rotation B. Optical density A. Sodium edetate C. Refractive index B. Potassium bromate D. Relative density C. Hydrochloric acid E. Distribution coefficient D. Potassium permanganate E. Sodium hydroxide 8. Which of the following antibiotics can be identified by means of maltol test? 3. An analytical chemist of a central industrial laboratory performs quantitative A. Streptomycin sulfate determination of penicillin sum in B. Doxycycline hydrochloride benzylpenicillin sodium salt by means of C. Amoxicillin iodometric method. What indicator is used? D. Lincomycin hydrochloride E. Kanamycin monosulfate A. Starch B. Phenolphthalein 9. A pharmaceutical analyst identifies C. Potassium chromate the substance of potassium acetate. What D. Methyl orange reagent confirms the presence of potassium E. Methyl red cation in the substance? 4. Paracetamol substance has been sent A. -

OBJ (Application/Pdf)
STUDIES OF CONJUGATED SYSTEMS* THE ATTEMPTED ADDITION OF THIOCYANOGEN TO 1,4-DIPHENYLBUTADIENE AND THE HYDROXYLATION OF REDUCTION PRODUCT OF l-PHENYLBUTADIENE-1,3 <r A THESIS SUBMITTED TO THE FACULTY OF ATLANTA UNIVERSITY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE BY WILLIAM GORDON DEWITT HENDERSON DEPARTMENT OF CHEMISTRY ATLANTA* GEORGIA JUNE 1948 ii TABLE OF CONTENTS Chapter Page I. INTRODUCTION 1 II. THEORETICAL 2 Part I 2 Part 9 III. EXPERIMENTAL. 15 Part I 15 Part II 18 17. CONCLUSIONS 21 BIBLIOGRAPHY 22 iii ACKNOWLEDGMENT My appreciation is hereby expressed to Mrs* K. A. Huggins and my relatives far their continuous encouragement which did much to inspire me to write this thesis* I am also grateful for the aid rendered me by Dr» H, C* MoBay and the Library Staff* I wish to express my sincere thanks to Dr* K* A* Huggins for his helpful suggestions and criticisms* Without his help and untiring guidance* and excellent courses* this work would not have been possible* CHAPTER I INTRODUCTION For many years investigators have been studying the reaotions of conjugated compounds* These compounds are of great theoretical im¬ portance in organic chemistry as veil as in industry* Sinoe the pos¬ tulation of the theory, of partial valenoy by. Thiele** the interest in conjugated compounds has steadily, increased* For some time workers in this laboratory have studied the reactions of the dienes with many reagents * The work herein reported is a continuation of such studies* This thesis is a report of a study whioh has been made of the aotion of thiooyanogen with diphenylbutadiene, hydrogenation of phenylbutadiene and the hycLroxlation of phenylbutene-2* Henry, "Unpublished Master’s Thesis", Atlanta University, (1957) 1 CHAPTER II THEORETICAL PART I Reaction of Thioeyanogen.—Numerous investigators have studied the reaotion of thioeyanogen with organic compounds* Thioeyanogen has been found to react both by substitution and by. -

Inorganic, Organic and Physical Chemistry- Iv
UNIT I HALOGEN FAMILY AND ZERO GROUP ELEMENTS TWO MARKS 1.What are halogens? Give an oxidation states of halogens. 2.What are interhalogens? 3.How chlorine monofluoride is prepared? 4.What are pseudohalogens?Give an example. 5.How thiocyanogen is prepared?What are its properties? 6.Write the characteristics of interhalogens. 7.What are noble gases? 8. Write the uses of xenon compound. INORGANIC, ORGANIC AND 9.How XeOF4 is prepared? PHYSICAL CHEMISTRY- IV 10.How XeF6 is prepared in laboratory? FIVE MARKS Subject Code: 18K4CH06 1.Explain the AX3 type of interhalogen compounds. 2.Explain the preparation and properties of IF7. 3.Explain the characteristics of interhalogen compounds. 4.Explain the preparation and properties of XeF4. 5. Explain the preparation and properties of cyanogens. 6.Explain the position of noble gases in the periodic table. TEN MARKS 1.Discuss the structures and uses of interhalogen compounds. 2.Describe the AX type of interhalogen compound. 3.Explain the comparative study of halogen compounds. 4.Describe the methods of preparation and properties of XeF6 and XeF2. HALOGEN FAMILY AND ZERO GROUP ELEMENTS contains two or more different halogen atoms (fluorine, chlorine, bromine, iodine, or astatine) and no atoms of elements from any other group. The halogens are a group in the periodic table consisting of five chemically Most interhalogen compounds known are binary (composed of only two distinct elements). related elements: fluorine (F), chlorine(Cl), bromine (Br), iodine (I), and astatine (At). The Their formulae are generally XYn, where n = 1, 3, 5 or 7, and X is the less electronegative of artificially created element 117, tennessine (Ts), may also be a halogen. -

United States Patent (19) 11 Patent Number: 4,910,263 Brois Et Al
United States Patent (19) 11 Patent Number: 4,910,263 Brois et al. (45) Date of Patent: Mar. 20, 1990 (54) OIL ADDITIVES CONTAINING A 3,382,215 5/1968 Baum ................. 525/351 X 3,647,849 3/1972 Venerable et al. 260/454 THIOCARBAMYL MOIETY 3,749,747 7/1973 Fancher .............................. 260/454 75) Inventors: Stanley J. Brois, Spring, Tex.; 3,867,360 2/1975 Jones ............................... 525/351 X Antonio Gutierrez, Mercerville, N.J. FOREIGN PATENT DOCUMENTS 73 Assignee: Exxon Research & Engineering Company, Florham Park, N.J. 514052 10/1939 United Kingdom .................. 252/47 (21) Appl. No.: 252,942 OTHER PUBLICATIONS E. Reid, "Organic Chemistry of Bivalent Sulfur', vol. (22 Filed: Oct. 3, 1988 VI, 1966, pp. 62-65 and 220-221. R. G. Guy et al., “Pseudohalogen Chemistry”, J. Chem. Related U.S. Application Data Soc. 1973, pp. 281-284. 60 Division of Ser. No. 89,449, Aug. 20, 1987, Pat. No. R. G. R. Bacon, Chapter 27, "Thiocyanates, Thi 4,794,146, which is a continuation of Ser. No. 908,786, ocyanogen, and Related Compounds,’ Organic Sulfur Sep. 18, 1986, Pat. No. 4,717,754, which is a continua tion of Ser. No. 776,019, Sep. 13, 1985, abandoned, Compounds, edited by N. Kharasch, vol. 1, Pergamon which is a continuation of Ser. No. 320,574, Nov. 21, Press, 1961, NY, Symposium Publications Division, pp. 1981, abandoned, which is a continuation of Ser. No. 306-325. 109,778, Jan. 7, 1980, abandoned. R. G. Guy, "Thiocyanogen Halides,", Mechanisms of 5ll Int. Cl." ................................................ C08F 8/32 Reactions of Sulfur Compounds, vol. -

Stanley Miller Papers
http://oac.cdlib.org/findaid/ark:/13030/kt300035nw No online items Stanley Miller Papers Special Collections & Archives, UC San Diego Special Collections & Archives, UC San Diego Copyright 2010 9500 Gilman Drive La Jolla 92093-0175 [email protected] URL: http://libraries.ucsd.edu/collections/sca/index.html Stanley Miller Papers MSS 0642 1 Descriptive Summary Languages: English Contributing Institution: Special Collections & Archives, UC San Diego 9500 Gilman Drive La Jolla 92093-0175 Title: Stanley Miller Papers Identifier/Call Number: MSS 0642 Physical Description: 92.7 Linear feet(227 archives boxes, 2 card file boxes, one flat box, and five map case folders) Date (inclusive): 1952-2010 Abstract: The papers of Stanley Miller, chemist, biologist, and professor at the University of California, San Diego, known for his research into the origins of life. The papers date from 1952 to 2010 and include correspondence, writing, referee reports, research and subject files, teaching materials, conference and meeting materials, grant proposals, and audio visual materials. Biography Stanley Miller was born in Oakland, California, in 1930. He received his bachelor's degree in chemistry from the University of California, Berkeley in 1951 and his Ph.D. from the University of Chicago in 1954. It was there, as a graduate student studying under Harold Urey, that Miller gained attention for his research into the origins of life. As part of his graduate research, Miller created an electric discharge experiment which showed how amino acids could be generated from chemicals thought to have been present on the primitive earth. Miller's results were published by the journal Science in 1953, attracting attention from both the scientific community and the popular press. -

Decomposition Products of Allyl Isothiocyanate in Aqueous Solutions
4584 J. Agric. Food Chem. 1997, 45, 4584−4588 Decomposition Products of Allyl Isothiocyanate in Aqueous Solutions Radomı´r Pecha´cˇek, Jan Velı´sˇek,* and Helena Hrabcova´ Department of Food Chemistry and Analysis, Institute of Chemical Technology, Technicka´ 1905, 166 28 Prague 6, Czech Republic Solutions containing 1000 mg kg-1 of allyl isothiocyanate (AITC) in buffers of pH 4, 6, and 8 were stored at 80 °C and analyzed for the content of AITC, its isomerization product allyl thiocyanate (ATC), and 9 other decomposition products. The major reaction products in alkaline buffers were allylamine (AA), allyl dithiocarbamate (ADTC), diallylthiourea (DATU), and carbon disulfide (CDS). CDS and AA also arose as the major products in buffers of pH 6 and 4. Newly identified decomposition products of AITC were ADTC, diallylurea (DAU), and diallyl sulfide (DAS). Mechanisms explaining the decomposition of AITC are presented and discussed. Keywords: Allyl isothiocyanate; degradation products; allyl thiocyanate; allylamine; diallylthiourea; diallylurea; allyl dithiocarbamate; allyl allyldithiocarbamate; allyl mercaptan; diallyl sulfide; diallyl disulfide; carbon disulfide; HPLC; GLC INTRODUCTION agrochemicals, their degradation products, and metabo- lites. Similarly, AITC is a toxic compound showing a Isothiocyanates (ITCs), also referred to as mustard wide range of biological activities. Owing to the elec- oils, have been found in a number of higher plants either trophilic character of the isothiocyanate functional in free form or as glucosinolates from which they are group, the toxicity is assumed to be associated with set free by enzymatic hydrolysis or by chemical conver- reactions of AITC with biological thiols, amines, and sion. alcohols in vivo. AITC (and its homologue methyl Allyl isothiocyanate (AITC) is one of the most wide- isothiocyanate) is used as a multipurpose soil fumigant spread isothiocyanates.