Thesis Reference
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Densifying Metal Hydrides with High Temperature and Pressure
3,784,682 United States Patent Office Patented Jan. 8, 1974 feet the true density. That is, by this method only theo- 3,784,682 retical or near theoretical densities can be obtained by DENSIFYING METAL HYDRIDES WITH HIGH making the material quite free from porosity (p. 354). TEMPERATURE AND PRESSURE The true density remains the same. Leonard M. NiebylsM, Birmingham, Mich., assignor to Ethyl Corporation, Richmond, Va. SUMMARY OF THE INVENTION No Drawing. Continuation-in-part of abandoned applica- tion Ser. No. 392,370, Aug. 24, 1964. This application The process of this invention provides a practical Apr. 9,1968, Ser. No. 721,135 method of increasing the true density of hydrides of Int. CI. COlb 6/00, 6/06 metals of Groups II-A, II-B, III-A and III-B of the U.S. CI. 423—645 8 Claims Periodic Table. More specifically, true densities of said 10 metal hydrides may be substantially increased by subject- ing a hydride to superatmospheric pressures at or above ABSTRACT OF THE DISCLOSURE fusion temperatures. When beryllium hydride is subjected A method of increasing the density of a hydride of a to this process, a material having a density of at least metal of Groups II-A, II-B, III-A and III-B of the 0.69 g./cc. is obtained. It may or may not be crystalline. Periodic Table which comprises subjecting a hydride to 15 a pressure of from about 50,000 p.s.i. to about 900,000 DESCRIPTION OF THE PREFERRED p.s.i. at or above the fusion temperature of the hydride; EMBODIMENT i.e., between about 65° C. -

The Identification of Olefins As Thiocyanates
THE IDENTIFICATION OF OLEFI NS AS THIOCYANATES 1 .. .SEP 2"/ i 938 THE IDENTIFICATION OF OLEFINS AS THIOCYANAT ES/ ( ' By George A. Dysinger I\ Bachelor of Science Oklahoma Agricultural and Mechanical College Stillwater, Oklahoma 1937 Submitted to the Department of Chemistry Oklahoma Agricultural and Mechanical College In partial fulfillme.nt of the requirements for the Degree of MASTER OF SCIENCE 1938 . ... .. '.'' .. ~ . ..... .. • • • • • • • • J • : ... ·:· .· ~- . .. ,. r f • • • - • • • • •• J • •• ; • • • ii !)fp 27 1938 APPROVED: HeadOttinm~- of : e Department of Onemistry ~~~e~ 108550 iii ACKNOWLEDGEMENT The author wishes to expres.s his sincere appreciation to Dr. o. c. Dermer under whose direction and with whose help this work has been done. He also wishes to acknowledge the assistance rendered by Everett L. Ada.ms in supplying apparatus and chemicals. iv TABLE OF CONTENTS Page Introduotion • . .. • . • • • 1 Historical Review. • . • • . • . 2 Experimental . • • • • . • . • . • • 6 Discussion of Results . .. ao Summary • • • • . • • • 22 Bibliography • • • • • • • • • • • • • • • • • • 23 Autobiography • • • • • . • • • • • • • • • . • 24 1 INTRODUCTION At present, the identification of low boiling unsatu rated hydrocarbons which yield only liquid addition products with the halogens and hydrogen halides is often a matter of considerable difficulty. This f'aot is sufficient reason for the study here described. In some cases, espeoially among terpen.es, the compounds formed by adding NOCl, 1203 , or »2o4 at one or more of the double bonds have been used as derivatives but these are i nconvenient to make,. of un certain composition, and decidedly unstable! This work con- '\, sists of (l) a'ttempts to find a convenient samll scale meth- od for adding (S0N)2 to olef1ns, (2) the determinations of the melting points of the derivatives, and (3) quantitatiye analysis of derivatives to prove their structure and purity. -

Inorganic Seminar Abstracts
C 1 « « « • .... * . i - : \ ! -M. • ~ . • ' •» »» IB .< L I B RA FLY OF THE. UN IVERSITY Of 1LLI NOIS 546 1^52-53 Return this book on or before the Latest Date stamped below. University of Illinois Library «r L161— H41 Digitized by the Internet Archive in 2012 with funding from University of Illinois Urbana-Champaign http://archive.org/details/inorganicsemi195253univ INORGANIC SEMINARS 1952 - 1953 TABLE OF CONTENTS 1952 - 1953 Page COMPOUNDS CONTAINING THE SILICON-SULFUR LINKAGE 1 Stanley Kirschner ANALYTICAL PROCEDURES USING ACETIC ACID AS A SOLVENT 5 Donald H . Wilkins THE SOLVENT PHOSPHORYL CHLORIDE, POCl 3 12 S.J. Gill METHODS FOR PREPARATION OF PURE SILICON 17 Alex Beresniewicz IMIDODISULFINAMIDE 21 G.R. Johnston FORCE CONSTANTS IN POLYATOMIC MOLECILES 28 Donn D. Darsow METATHESIS IN LIQUID ARSENIC TRICHLORIDE 32 Harold H. Matsuguma THE RHENI DE OXIDATION STATE 40 Robert L. Rebertus HALOGEN CATIONS 45 L.H. Diamond REACTIONS OF THE NITROSYL ION 50 M.K. Snyder THE OCCURRENCE OF MAXIMUM OXIDATION STATES AMONG THE FLUOROCOMPLEXES OF THE FIRST TRANSITION SERIES 56 D.H. Busch POLY- and METAPHOSPHATES 62 V.D. Aftandilian PRODUCTION OF SILICON CHLORIDES BY ELECTRICAL DISCHARGE AND HIGH TEMPERATURE TECHNIQUES 67 VI. £, Cooley FLUORINE CONTAINING OXYHALIDES OF SULFUR 72 E.H. Grahn PREPARATION AND PROPERTIES OF URANYL CARBONATES 76 Richard *• Rowe THE NATURE OF IODINE SOLUTIONS 80 Ervin c olton SOME REACTIONS OF OZONE 84 Barbara H. Weil ' HYDRAZINE BY ELECTROLYSIS IN LIQUID AMMONIA 89 Robert N. Hammer NAPHTHAZARIN COMPLEXES OF THORIUM AND RARE EARTH METAL IONS 93 Melvin Tecotzky THESIS REPORT 97 Perry Kippur ION-PAIR FORMATION IN ACETIC ACID 101 M.M. -

Extraction of Cyanides from Waste Solutions of Cyanidation of .Lotation
Chemistry for Sustainable Development 12 (2004) 431436 431 Extraction of Cyanides from Waste Solutions of Cyanidation of lotation Concentrates from Kholbinskoye Gold Deposit A. A. KOCHANOV1, A. A. RYAZANTSEV1, A. A. BATOEVA2, D. B. ZHALSANOVA2, A. M. BADALYAN3 and O. V. POLYAKOV3 1Siberian Transport University, Ul. D. Kovalchuk 191, Novosibirsk 630049 (Russia) E-mail: [email protected] 2Baikal Institute of Nature Management, Siberian Branch of the Russian Academy of Sciences, Ul. Sakhyanovoy 6, Ulan Ude 670047 (Russia) 3A. V. Nikolaev Institute of Inorganic Chemistry, Siberian Branch of the Russian Academy of Sciences, Pr. Akademika Lavrentyeva 3, Novosibirsk 630090 (Russia) (Received April 21, 2003; in revised form January 6, 2004) Abstract Processes that occur during extraction of cyanides from cyanidation solutions using centrifugal bubbling apparatuses (CBA) as reactors have been studied. In the eddy chamber of CBA (ðÍ < 3), one can observe virtually complete removal of HCN from solution and precipitation of heavy metals in the form of insoluble x - 1 compounds. The electronic absorption spectra of solutions treated in CBA suggest that destruction of [Cu(CN)x] , + 2+ 2 oxidation of Cu to Cu by air oxygen, and oxidation of thiocyanates in the presence of SO23, forming 2 HCN and SO4 , are also accompanied by the emergence of stable intermediate products (SCN)2 and (SCN)x of oxidation in solution. INTRODUCTION formed after acidification of the waste process- ing solutions of cyanidation to ðÍ 62.5 fol- Cyanides are widely used in extraction of lowed by absorption of HCN by alkaline solu- precious metals, mainly fine gold and silver, tions. -

(12) United States Patent (10) Patent No.: US 7,732,534 B2 Luo Et Al
US007732534B2 (12) United States Patent (10) Patent No.: US 7,732,534 B2 Luo et al. (45) Date of Patent: Jun. 8, 2010 (54) POLYMERS FUNCTIONALIZED WITH 6,172,160 B1 1/2001 Nakamura et al. NITRO COMPOUNDS 6, 194505 B1 2/2001 Sone et al. .................. 524/432 6, 197,888 B1 3/2001 Luo ........................... 525,247 (75) Inventors: Steven Luo, Copley, OH (US); Ryuji Nakagawa, Tokyo (JP) 6.255.416 B1 7/2001 Sone et al. .................. 526,153 6.271,315 B1* 8/2001 Kiessling et al. ......... 525,326.1 (73) Assignee: Bridgestone Corporation (JP) 6,291,591 B1 9/2001 Luo ........................... 525, 191 6,303,692 B1 10/2001 Luo ........................... 525, 191 (*) Notice: Subject to any disclaimer, the term of this 6,699,813 B2 3/2004 Luo et al. ................... 502,119 patent is extended or adjusted under 35 6,759,497 B2 7/2004 Grun et al. U.S.C. 154(b) by 526 days. 6,838,526 B1 1/2005 Sone et al. (21) Appl. No.: 11/710,845 6,897,270 B2 5/2005 Ozawa et al. ................. 526,88 6,977.281 B1* 12/2005 Ozawa et al. ............... 525/377 (22) Filed: Feb. 26, 2007 6,992,147 B1 1/2006 Ozawa et al. ............... 525,342 7,008,899 B2 3/2006 Luo et al. ................... 502,131 Prior Publication Data (65) 7,094,849 B2 8/2006 Luo et al. ................... 526, 164 US 2008/OO51552 A1 Feb. 28, 2008 7,351,776 B2 4/2008 Tartamella et al. Related U.S. Application Data 2004/O147694 A1 7/2004 Sone et al. -

United States Patent Office Patented Feb
3,644,463 United States Patent Office Patented Feb. 22, 1972 2 agent that is introduced. Thus, for example, good yields 3,644,463 of 1,2-vinylene-bisthiocyanate are obtained by adding PRODUCTION OF ALEPHATIC 1,2-BISTHIOCYANATES acetylene gas, while the addition of ethylene results in the Richard Parke Welcher, Old Greenwich, Conn., assignor production of 1,2-dithiocyanoethane. The corresponding to American Cyanamid Company, Stamford, Conn. monoalkyl-substituted vinylene bisthiocyanates are pro No Drawing. Filed May 15, 1968, Ser. No. 729,375 duced when monoalkyl acetylene containing an alkyl radi Int, C. C07c 161/02 cal of from 1 to 16 carbn atoms are used, such as methyl U.S. C. 260-454 5 Claims acetylene, heptyne, octyne, octadecyne, and the like. Monoalkyl-dithiocyanoethanes are produced in similar O manner when monoalkylethylenes containing alkyl radi cals of 1-16 carbon atoms are used; typical of these are ABSTRACT OF THE DISCLOSURE propylene, isobutylene, octylethylene, hexadecyl-ethylene Aliphatic 1,2-bisthiocyanates are produced by first pre and the like. Dialkyl ethylenes may likewise be used, the paring a solution of thiocyanogen in a water-insoluble preferred reagents being those wherein the two alkyl sub liquid organic solvent such as toluene having an aqueous stituents taken together have a total of from 2 to 16 car solution of an inorganic halide admixed therewith, draw bon atoms. Aryl-substituted olefins such as styrene may ing off the aqueous phase, adding an alpha-olefin or acetyl also be used. The principles of the invention can also ene and reacting at a temperature below about 20° C. -
![Chemistry Assessment Unit AS 1 Assessing Basic Concepts in Physical and Inorganic Chemistry AC112 [AC112]](https://docslib.b-cdn.net/cover/7809/chemistry-assessment-unit-as-1-assessing-basic-concepts-in-physical-and-inorganic-chemistry-ac112-ac112-3727809.webp)
Chemistry Assessment Unit AS 1 Assessing Basic Concepts in Physical and Inorganic Chemistry AC112 [AC112]
Centre Number 71 Candidate Number ADVANCED SUBSIDIARY (AS) General Certificate of Education 2011 Chemistry Assessment Unit AS 1 assessing Basic Concepts in Physical and Inorganic Chemistry AC112 [AC112] WEDNESDAY 15 JUNE, AFTERNOON TIME 1 hour 30 minutes. INSTRUCTIONS TO CANDIDATES Write your Centre Number and Candidate Number in the spaces provided at the top of this page. For Examiner’s Answer all sixteen questions. use only all ten Section A Question Answer questions in . Record your answers by Marks marking the appropriate letter on the answer sheet provided. Use only Number the spaces numbered 1 to 10. Keep in sequence when answering. Section A Answer all six questions in Section B. Write your answers in the 1-10 spaces provided in this question paper. Section B INFORMATION FOR CANDIDATES 11 12 The total mark for this paper is 100. Quality of written communication will be assessed in question 15(f). 13 In Section A all questions carry equal marks, i.e. two marks 14 for each question. In Section B the figures in brackets printed down the right-hand 15 side of pages indicate the marks awarded to each question or part 16 question. 1 1 1 2 9 2 A Periodic Table of Elements (including some data) is provided. Total Marks 6898 Section A Foreachofthefollowingquestionsonlyoneoftheletteredresponses(A–D)iscorrect. Select the correct response in each case and mark its code letter by connecting the dots as illustrated on the answer sheet. 1 Anelementwhichformsanionsmallerthanitsatomis A chlorine. -

WSU Tri-Cities Laboratory Safety Manual
WSU Tri-Cities Laboratory Safety Manual December 2014 Contents Section I - Introduction ................................................................................................................... 1 A. Purpose ............................................................................................................................. 1 B. Scope and Application ..................................................................................................... 1 C. Implementation of the Chemical Hygiene Plan (CHP) .................................................... 2 D. Responsibilities ................................................................................................................ 3 Section II – General Policies and Recommendations ..................................................................... 5 A. Basic Rules and Procedures ............................................................................................. 5 B. Chemical Procurement, Storage, and Distribution .......................................................... 6 C. Exposure Monitoring ....................................................................................................... 8 D. Housekeeping ................................................................................................................. 10 E. Medical Surveillance ..................................................................................................... 10 F. Personal Protective Equipment ..................................................................................... -
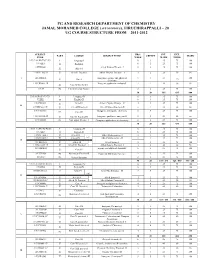
UG Syllabus.Pdf
PG AND RESEARCH DEPARTMENT OF CHEMISTRY JAMAL MOHAMED COLLEGE (AUTONOMOUS), TIRUCHIRAPPALLI – 20 UG COURSE STRUCTURE FROM 2011-2012 SUBJECT HRS / INT. EXT. ` PART COURSE SUBJECT TITLE CREDIT MARK CODE WEEK MARK MARK 11U1LA1/H1/T1/F1/U1 I Language-I 6 3 25 75 100 11UILE1 6 3 25 75 100 II English-I 11UPH1301 Allied Physics Theory - I 5 3 25 75 100 III Allied-I 11UPH1302:1P III Allied-I-Practical Allied Physics Practical – I 3 2 20 30 50 11UCH1401 Inorganic, organic and physical 5 5 25 100 I III Core-I 75 chemistry 11UCH1402:1P Inorganic qualitative analysis-I 3 2 20 30 50 III Core-II-Practical-I 11U19 IV Environmental Studies 2 2 25 75 100 30 20 165 435 600 U2LA2/H2/T2/F2/U2 I Language-II 6 3 25 75 100 U2LE2 English-II 6 3 25 75 100 II 11UPH2303 III Allied-II Allied Physics Theory – II 4 3 25 75 100 II 11UPH2302:2P III Allied-II Practical Allied Physics Practical-II 3 2 20 30 50 11UCH2403 Inorganic and organic chemistry 6 5 25 75 100 III Core-III 11UCH2402:2P III Core-II Practical-II Inorganic qualitative analysis-II 3 2 20 30 50 11UCH2601 IV Non-major Elective-I Computer applications in chemistry 2 2 25 75 100 30 20 165 435 600 11U3LA3/H3/T3/F3/U3 I Language-III 6 3 25 75 100 11U3LE3 II English-III 6 3 25 75 100 11UMA3304:2 III Allied-III Allied Mathematics - I 5 3 25 75 100 (or) 11UMA3305:2 III Allied-1V Allied Mathematics - II 3 2 25 75 100 III 11UBO3304 III Allied-III Allied Botany-I 5 3 25 75 100 11UBO3305:1P III Allied-III Practical Allied Botany Practical-I 3 2 20 30 50 11UCH3404 organic and physical chemistry 6 5 25 75 100 III Core-IV -

Nomenclature of Inorganic Chemistry (IUPAC Recommendations 2005)
NOMENCLATURE OF INORGANIC CHEMISTRY IUPAC Recommendations 2005 IUPAC Periodic Table of the Elements 118 1 2 21314151617 H He 3 4 5 6 7 8 9 10 Li Be B C N O F Ne 11 12 13 14 15 16 17 18 3456 78910 11 12 Na Mg Al Si P S Cl Ar 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe 55 56 * 57− 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 Cs Ba lanthanoids Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn 87 88 ‡ 89− 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 Fr Ra actinoids Rf Db Sg Bh Hs Mt Ds Rg Uub Uut Uuq Uup Uuh Uus Uuo * 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu ‡ 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Ac Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr International Union of Pure and Applied Chemistry Nomenclature of Inorganic Chemistry IUPAC RECOMMENDATIONS 2005 Issued by the Division of Chemical Nomenclature and Structure Representation in collaboration with the Division of Inorganic Chemistry Prepared for publication by Neil G. -
![Chemistry Assessment Unit AS 1 Assessing Basic Concepts in Physical and Inorganic Chemistry AC112 [AC112]](https://docslib.b-cdn.net/cover/1972/chemistry-assessment-unit-as-1-assessing-basic-concepts-in-physical-and-inorganic-chemistry-ac112-ac112-5331972.webp)
Chemistry Assessment Unit AS 1 Assessing Basic Concepts in Physical and Inorganic Chemistry AC112 [AC112]
Centre Number 71 Candidate Number ADVANCED SUBSIDIARY (AS) General Certificate of Education 2011 Chemistry Assessment Unit AS 1 assessing Basic Concepts in Physical and Inorganic Chemistry AC112 [AC112] WEDNESDAY 15 JUNE, AFTERNOON TIME 1 hour 30 minutes. INSTRUCTIONS TO CANDIDATES Write your Centre Number and Candidate Number in the spaces provided at the top of this page. For Examiner’s Answer all sixteen questions. use only all ten Section A Question Answer questions in . Record your answers by Marks marking the appropriate letter on the answer sheet provided. Use only Number the spaces numbered 1 to 10. Keep in sequence when answering. Section A Answer all six questions in Section B. Write your answers in the 1-10 spaces provided in this question paper. Section B INFORMATION FOR CANDIDATES 11 12 The total mark for this paper is 100. Quality of written communication will be assessed in question 15(f). 13 In Section A all questions carry equal marks, i.e. two marks 14 for each question. In Section B the figures in brackets printed down the right-hand 15 side of pages indicate the marks awarded to each question or part 16 question. 1 1 1 2 9 2 A Periodic Table of Elements (including some data) is provided. Total Marks 6898 Section A Foreachofthefollowingquestionsonlyoneoftheletteredresponses(A–D)iscorrect. Select the correct response in each case and mark its code letter by connecting the dots as illustrated on the answer sheet. 1 Anelementwhichformsanionsmallerthanitsatomis A chlorine. -

Vii. Publications(April 2011-March 2012) (Pdf)
VII. PUBLICATIONS (APRIL 2011 – MARCH 2012) VII. PUBLICATIONS Since 1967, the institute published the Annual Report of the KURRI, containing the original papers written by researchers of the institute and users of other organizations. It played an important role in showing the activities of the institute. However, due to a variety of research fields covered by researchers in the institute, it became difficult to contain all the original papers. Therefore, the Annual Report was discontinued in 1995. The Progress Report of the KURRI has been issued from 1991 in English and summarizes the abstracts of the published papers, reviews, book titles and current research activities of the KURRI. The Technical Report of the KURRI (KURRI-TR) has been issued occasionally from 1965. It contains technical data in Japanese with English summaries. The KUR Report (KURRI-KR), issued in Japanese since 1996, is the proceedings of symposia and technical meetings held at the institute. Furthermore, the CD-ROM version (KURRI-KR(CD)) has been issued from 2004. The KUR Report (KURRI-KR) KURRI-KR-165 Meeting on the Future Project of the Kyoto University Research Reactor Institute (2011) KURRI-KR-166 Specialists' Meeting on the Chemistry and Technology of Actinide Elements 2010 (2011) KURRI-KR-167 Proceedings of the Specialists' Meeting on Radioactive Waste Management 2011 (2012) KURRI-KR-168 Proceedings of the Specialist Research Meeting on Science and Engineering of Unstable Nuclei and Their Uses on Condensed Matter Physics (2012) KURRI-KR-169 Proceedings of the