Complex Metal Halides in Fused Alkali Nitrates Marlowe L
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Attachment 3-1 Guidance for Developing Ecological Soil
Attachment 3-1 Guidance for Developing Ecological Soil Screening Levels (Eco-SSLs) Eco-SSL Standard Operating Procedure (SOP # 1): Plant and Soil Invertebrate Literature Search and Acquisition OSWER Directive 92857-55 November 2003 This page intentionally left blank OVERVIEW Currently, there is a lack of clear guidance in setting terrestrial effect thresholds when conducting risk assessments. Without an EPA-approved, peer-reviewed, ecologically-based terrestrial effect database, the process to develop thresholds is problematic both to EPA, other federal agencies, states, and concerned private parties. Identification of published toxicity studies on invertebrates, microbial processes and plants is a key step in the derivation of benchmarks. The purpose of the Task Group 4, Standard Operating Procedure Number 1: Literature Search and Acquisition (referred to as TG4-SOP#1) is to document procedures used to identify and acquire potentially relevant toxicology literature for use in setting ecological soil screening levels. The literature search strategy is designed to locate worldwide terrestrial toxicity literature that includes the effects of chemicals of concern on terrestrial soil-dwelling invertebrates and plants. The literature acquisition process is designed to ensure timely acquisition of relevant publications. LITERATURE IDENTIFICATION Potentially relevant literature for developing ecological soil screening levels (Eco-SSLs) is identified by examining hard copies of relevant journals, bibliographies and guidance publications and through the use of a comprehensive computerized literature search strategy. These procedures are designed to locate worldwide terrestrial toxicology literature that includes the effects of specific toxic substances with an emphasis on exposure via soil. Paper-based Literature Identification The paper-based literature identification process includes the scanning of relevant review article bibliographies and key journals held in the U.S. -
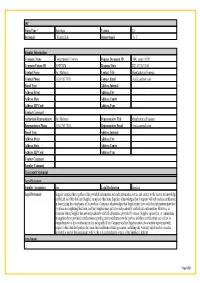
Material Composition Declaration
IPC Form Type * Distribute Version 2.0 Sectionals - MaterialInfo Subsectionals - A- C Supplier Information Company Name Contemporary Controls Request Document ID EISK_series_MCD Company Unique ID 363857474 Response Date 2021-07-30-12:00 Contact Name Neil Maloney Contact Title Manufacturing Engineer Contact Phone (630) 963 7070; Contact Email [email protected] Email Type Address Internal Address Street Address City Address State Address Contry Address ZIP Code Address Type Contact Comment Authorized Representative Neil Maloney Representative Title Manufacturing Engineer Representative Phone (630) 963 7070; Representative Email [email protected] Email Type Address Internal Address Street Address City Address State Address Contry Address ZIP Code Address Type Contact Comment Supplier Comment Uncertainty Statement Legal Statement Supplier Acceptance true Legal Declaration Standard Legal Statement Supplier certifies that it gathered the provided information and such information is true and correct to the best of its knowledge and belief, as of the date that Supplier completes this form. Supplier acknowledges that Company will rely on this certification in determining the compliance of its products. Company acknowledges that Supplier may have relied on information provided by others in completing this form, and that Supplier may not have independently verified such information. However, in situations where Supplier has not independently verified information provided by others, Supplier agrees that, at a minimum, its suppliers have provided certifications regarding their contributions to the part(s), and those certifications are at least as comprehensive as the certification in this paragraph. If the Company and the Supplier enter into a written agreement with respect to the identified part(s), the terms and conditions of that agreement, including any warranty rights and/or remedies provided as part of that agreement, will be the sole and exclusive source of the Supplier’s liability Attachment . -

20210311 IAEG AD-DSL V5.0 for Pdf.Xlsx
IAEGTM AD-DSL Release Version 4.1 12-30-2020 Authority: IAEG Identity: AD-DSL Version number: 4.1 Issue Date: 2020-12-30 Key Yellow shading indicates AD-DSL family group entries, which can be expanded to display a non-exhaustive list of secondary CAS numbers belonging to the family group Substance Identification Change Log IAEG Regulatory Date First Parent Group IAEG ID CAS EC Name Synonyms Revision Date ECHA ID Entry Type Criteria Added IAEG ID IAEG000001 1327-53-3 215-481-4 Diarsenic trioxide Arsenic trioxide R1;R2;D1 2015-03-17 2015-03-17 100.014.075 Substance Direct Entry IAEG000002 1303-28-2 215-116-9 Diarsenic pentaoxide Arsenic pentoxide; Arsenic oxide R1;R2;D1 2015-03-17 2015-03-17 100.013.743 Substance Direct Entry IAEG000003 15606-95-8 427-700-2 Triethyl arsenate R1;R2;D1 2015-03-17 2017-08-14 100.102.611 Substance Direct Entry IAEG000004 7778-39-4 231-901-9 Arsenic acid R1;R2;D1 2015-03-17 2015-03-17 100.029.001 Substance Direct Entry IAEG000005 3687-31-8 222-979-5 Trilead diarsenate R1;R2;D1 2015-03-17 2017-08-14 100.020.890 Substance Direct Entry IAEG000006 7778-44-1 231-904-5 Calcium arsenate R1;R2;D1 2015-03-17 2017-08-14 100.029.003 Substance Direct Entry IAEG000009 12006-15-4 234-484-1 Cadmium arsenide Tricadmium diarsenide R1;R2;D1 2017-08-14 2017-08-14 Substance Direct Entry IAEG000021 7440-41-7 231-150-7 Beryllium (Be) R2 2015-03-17 2019-01-24 Substance Direct Entry IAEG000022 1306-19-0 215-146-2 Cadmium oxide R1;R2;D1 2015-03-17 2017-08-14 100.013.770 Substance Direct Entry IAEG000023 10108-64-2 233-296-7 Cadmium -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -
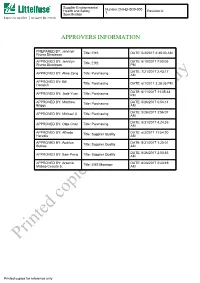
Printed Copies for Reference Only
Supplier Environmental Health and Safety Number:CHI-EHS30-000 Revision:A Specification 1 APPROVERS INFORMATION PREPARED BY: Jennilyn Rivera Dinglasan Title: EHS DATE: 5/3/2017 2:35:30 AM APPROVED BY: Jennilyn DATE: 8/10/2017 7:00:05 Rivera Dinglasan Title: EHS PM DATE: 7/21/2017 2:43:17 APPROVED BY: Aline Zeng Title: Purchasing AM APPROVED BY: Bill Hemrich Title: Purchasing DATE: 6/1/2017 3:28:35 PM DATE: 6/11/2017 11:05:48 APPROVED BY: Jade Yuan Title: Purchasing PM APPROVED BY: Matthew DATE: 5/26/2017 6:54:14 Briggs Title: Purchasing AM DATE: 5/26/2017 3:56:07 APPROVED BY: Michael Ji Title: Purchasing AM DATE: 8/31/2017 4:24:35 APPROVED BY: Olga Chen Title: Purchasing AM APPROVED BY: Alfredo DATE: 6/2/2017 11:54:20 Heredia Title: Supplier Quality AM APPROVED BY: Audrius DATE: 5/31/2017 1:20:01 Sutkus Title: Supplier Quality AM DATE: 5/26/2017 2:03:53 APPROVED BY: Sam Peng Title: Supplier Quality AM APPROVED BY: Arsenio DATE: 6/28/2017 2:43:49 Mabao Cesista Jr. Title: EHS Manager AM Printed copies for reference only Printed copies for reference only Supplier Environmental Health and Safety Number:CHI-EHS30-000 Revision:A Specification 1 1.0 Purpose and Scope 1.1 This specification provides general requirements to suppliers regarding Littelfuse Inc’s EHS specification with regards to regulatory compliance, EHS management systems, banned and restricted substances, packaging, and product environmental content reporting. 1.2 This specification applies to all equipment, materials, parts, components, packaging, or products supplied to Littelfuse, Inc. -

Draft Chemicals (Management and Safety) Rules, 20Xx
Draft Chemicals (Management and Safety) Rules, 20xx In exercise of the powers conferred by Sections 3, 6 and 25 of the Environment (Protection) Act, 1986 (29 of 1986), and in supersession of the Manufacture, Storage and Import of Hazardous Chemical Rules, 1989 and the Chemical Accidents (Emergency Planning, Preparedness and Response) Rules, 1996, except things done or omitted to be done before such supersession, the Central Government hereby makes the following Rules relating to the management and safety of chemicals, namely: 1. Short Title and Commencement (1) These Rules may be called the Chemicals (Management and Safety) Rules, 20xx. (2) These Rules shall come into force on the date of their publication in the Official Gazette. Chapter I Definitions, Objectives and Scope 2. Definitions (1) In these Rules, unless the context otherwise requires (a) “Act” means the Environment (Protection) Act, 1986 (29 of 1986) as amended from time to time; (b) “Article” means any object whose function is determined by its shape, surface or design to a greater degree than its chemical composition; (c) “Authorised Representative” means a natural or juristic person in India who is authorised by a foreign Manufacturer under Rule 6(2); (d) “Chemical Accident” means an accident involving a sudden or unintended occurrence while handling any Hazardous Chemical, resulting in exposure (continuous, intermittent or repeated) to the Hazardous Chemical causing death or injury to any person or damage to any property, but does not include an accident by reason only -

Compound Name
Indicate Type of Compound: Compound Name Write your answer here I = ionic, A= acid, M = molecular manganese (II) bromite I Mn(BrO2)2 manganese (II) phosphite I Mn3(PO3)2 rubidium sulfite I Rb2SO3 hydroselenic acid A H2Se(aq) sodium perbromate I NaBrO4 cobalt (III) chromate I Co2(CrO4)3 antimony (V) nitrite I Sb(NO2)5 chloric acid A HClO3(aq) pentaselenium decabromide M Se5Br10 disulfur decachloride M S2Cl10 nickel (III) nitrate I Ni(NO3)3 copper (II) bromide I CuBr2 nickel (II) hydrogen phosphate I NiHPO4 iron (II) hydrogen sulfate I Fe(HSO4)2 bismuth (V) acetate I Bi(C2H3O2)5 sulfurous acid A H2SO3(aq) sulfuric acid A H2SO4(aq) nickel (II) chloride I NiCl2 tin (IV) phosphate I Sn3(PO4)4 mercury (I) iodate I Hg2(IO3)2 Compound Indicate Type of Compound: Write your answer here Formula I = ionic, A= acid, M = molecular Co(HCO3)2 I (with VOS metal) cobalt (II) hydrogen carbonate Cs2S I cesium sulfide Ca(IO2)2 I calcium iodite Ba2C I barium carbide Mn(CO3)2 I (with VOS metal) manganese (IV) carbonate CuBrO2 I (with VOS metal) copper (I) bromite AgHS I silver hydrogen sulfide C9N10 M nonacarbon decanitride CrI2 I (with VOS metal) chromium (II) iodide Mg(NO3)2 I magnesium nitrate HC2H3O2 (aq) A acetic acid HClO2 (aq) A chlorous acid Be(IO4)2 I beryllium periodate HIO4(aq) A periodic acid BaO I barium oxide Cd(BrO3)2 I cadmium bromate Bi(CN)5 I (with VOS metal) bismuth (V) cyanide AuHS I (with VOS metal) gold (I) hydrogen sulfide AuClO I (with VOS metal) gold (I) hypochlorite Na2CO3 I sodium carbonate Indicate Type of Compound: Compound Name -

Validation of HVOF WC/Co Thermal Spray Coatings As a Replacement for Hard Chrome Plating on Aircraft Landing Gear
Naval Research Laboratory Washington, DC 20375-5320 NRL/MR/6170--04-8762 Validation of HVOF WC/Co Thermal Spray Coatings as a Replacement for Hard Chrome Plating on Aircraft Landing Gear BRUCE D. SARTWELL Surface Chemistry Branch, Chemistry Division KEITH O. LEGG Rowan Technology Group, Libertyville, IL JERRY SCHELL GE Aircraft Engines, Cincinnati, OH JOHN SAUER Sauer Engineering, Cincinnati, OH PAUL NATISHAN Center for Corrosion Science, Chemistry Division DENNIS DULL AND JOHN FALKOWSKI Boeing Corporation, Seattle, WA PHILIP BRETZ Metcut Research, Cincinnati, OH JON DEVEREAUX Naval Aviation Depot, Jacksonville, FL CRAIG EDWARDS General Atomics, Ogden, UT DONALD PARKER NASA Kennedy Space Center, Cape Canaveral, FL March 31, 2004 Approved for public release: distribution is unlimited. TABLE OF CONTENTS Table of Contents................................................................................................................iii List of Figures.....................................................................................................................ix List of Tables..................................................................................................................... xv List of Acronyms ............................................................................................................. xix Acknowledgments............................................................................................................ xxi 1. Background................................................................................................................1 -

Nited States Patent O?Ice Patented Nov
3,007,818 nited States Patent O?ice Patented Nov. 7, 1961 1 2 3,007,818 which often impairs the operation of certain types of PROTECTIVE COATINGS 0N METALS equipment such as relays and switches. The chromate Kurt Erich Schimkus, Chicago, Ill., assignor to Acme coatings referred to have been employed in an effort to Steel Company, Chicago, 111., a corporation of Illinois overcome this di?iculty. No Drawing. Filed Mar. 31, 1958, Ser. No. 724,794 It has been the usual practice to apply chromate coat 44 Claims. (Cl. 148-616) ings by an immersion process although electrolytic meth ods have sometimes been employed. The compositions This invention relates to protective coatings on metals of these baths are largely made up according to secret and to coating baths and coating methods for applying formulae but it is known that they all contain two basic the coatings. ' ingredients, viz., hexavalent chromium ions and mineral Many coatings and coating methods have been utilized acid with the addition, in some cases, of one or more heretofore to protect base metals, i.e., metals which cor organic acids. On immersion, the zinc coated or cad rode and discolor upon exposure to high humidity condi mium plated article is attacked by the mineral acid with tions and to salt water and the like. In certain instances an attendant rise in the pH of the solution adjacent to it is desirable that such coatings be transparent whereby 15 the metal. At the same time some hexavalent chromium to leave substantially unchanged the appearance of the is reduced to the trivalent state. -

Potential Chemical Contaminants in the Marine Environment
Potential chemical contaminants in the marine environment An overview of main contaminant lists Victoria Tornero, Georg Hanke 2017 EUR 28925 EN This publication is a Technical report by the Joint Research Centre (JRC), the European Commission’s science and knowledge service. It aims to provide evidence-based scientific support to the European policymaking process. The scientific output expressed does not imply a policy position of the European Commission. Neither the European Commission nor any person acting on behalf of the Commission is responsible for the use that might be made of this publication. Contact information Name: Victoria Tornero Address: European Commission Joint Research Centre, Directorate D Sustainable Resources, Water and Marine Resources Unit, Via Enrico Fermi 2749, I-21027 Ispra (VA) Email: [email protected] Tel.: +39-0332-785984 JRC Science Hub https://ec.europa.eu/jrc JRC 108964 EUR 28925 EN PDF ISBN 978-92-79-77045-6 ISSN 1831-9424 doi:10.2760/337288 Luxembourg: Publications Office of the European Union, 2017 © European Union, 2017 The reuse of the document is authorised, provided the source is acknowledged and the original meaning or message of the texts are not distorted. The European Commission shall not be held liable for any consequences stemming from the reuse. How to cite this report: Tornero V, Hanke G. Potential chemical contaminants in the marine environment: An overview of main contaminant lists. ISBN 978-92-79-77045-6, EUR 28925, doi:10.2760/337288 All images © European Union 2017 Contents Acknowledgements ................................................................................................ 1 Abstract ............................................................................................................... 2 1 Introduction ...................................................................................................... 3 2 Compilation of substances of environmental concern ............................................. -

Request for Building Environmentally Hazardous Substance Control System
[Attachment 1] Request for building environmentally hazardous substance control system The suppliers shall control containing in or adhesion to products to be supplied to NTN of materials specified in “I. NTN-prohibited substances,” “II. NTN observation-requisite substances,” and “III. Substances to be reported as requested by NTN” in the list of NTN environmentally hazardous substances shown in Attachment 4 and build the reporting (to NTN) and assurance system (especially, certification for nonuse of ELV/RoHS 10 substances). The requirements are shown below. The suppliers can make reference to guidelines for environmentally hazardous substance control published by “Joint Article Management Promotion-consortium (JAMP)” (available from our website). 1 Control of upstream end 1.1 Supply chain [1] Make a list of parts, materials, sub-materials, and packaging materials (hereinafter, simply referred to as parts, etc. and packaging materials) composing the products to be supplied to NTN and their suppliers for control. [2] Trace the supply chain of parts, etc. and packaging materials back to raw material manufacturers and make a list of them for control. [3] Provide your environmentally hazardous substance control criteria to your suppliers, request them to conform to the criteria, and understand the status of environmentally hazardous substance control performed by your suppliers by means of audit or with reference to submitted check sheets. 1.2 Recognition of information on substances contained in parts, etc. and packaging materials [1] Obtain composition tables, analytical data, mill sheets, SDS, etc. for parts, etc. and packaging materials from your suppliers or request your suppliers to conduct an investigation to recognize information on environmentally hazardous substances contained in products to be supplied. -
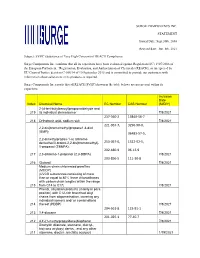
SURGE COMPONENTS INC. STATEMENT Issued Date: Sept.30Th, 2016 Revised Date: Jun. 8Th, 2021 Subject: SVHC (Substance of Very High
SURGE COMPONENTS INC. STATEMENT Issued Date: Sept.30th, 2016 Revised date: Jun. 8th, 2021 Subject: SVHC (Substance of Very High Concern) of REACH Compliance Surge Components Inc. confirms that all its capacitors have been evaluated against Regulation (EC) 1907/2006 of the European Parliament, “Registration, Evaluation, and Authorization of Chemicals (REACH), as interpreted by EU Court of Justice decision C-106/14 of 10 September 2015 and is committed to provide our customers with information about substances in its products as required. Surge Components Inc.reports that all REACH SVHC(shown in the table below) are not present within its capacitors. Inclusion Date Index Chemical Name EC Number CAS Number (M/D/Y) 2-(4-tert-butylbenzyl)propionaldehyde and 219 its individual stereoisomer 7/8/2021 237-560-2 13840-56-7 218 Orthoboric acid, sodium salt 7/8/2021 221-967-7, 3296-90-0, 2,2-bis(bromomethyl)propane1,3-diol (BMP); 36483-57-5, 2,2-dimethylpropan-1-ol, tribromo derivative/3-bromo-2,2-bis(bromomethyl)- 253-057-0, 1522-92-5, 1-propanol (TBNPA); 202-480-9 96-13-9 217 2,3-dibromo-1-propanol (2,3-DBPA) 7/8/2021 203-856-5 111-30-8 216 Glutaral 7/8/2021 Medium-chain chlorinated paraffins (MCCP) (UVCB substances consisting of more than or equal to 80% linear chloroalkanes with carbon chain lengths within the range 215 from C14 to C17) 7/8/2021 Phenol, alkylation products (mainly in para position) with C12-rich branched alkyl chains from oligomerisation, covering any individual isomers and/ or combinations 214 thereof (PDDP) 7/8/2021 204-661-8 123-91-1 213 1,4-dioxane 7/8/2021 201-025-1 77-40-7 212 4,4'-(1-methylpropylidene)bisphenol 7/8/2021 Dioctyltin dilaurate, stannane, dioctyl-, bis(coco acyloxy) derivs., and any other 211 stannane, dioctyl-, bis(fatty acyloxy) 1/19/2021 derivs.