The Metal–Ligand Electronic Parameter (MLEP)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Geoffrey Wilkinson
THE LONG SEARCH FOR STABLE TRANSITION METAL ALKYLS Nobel Lecture, December 11, 1973 by G EOFFREY W ILKINSON Imperial College of Science & Technology, London, England Chemical compounds in which there is a single bond between a saturated car- bon atom and a transition metal atom are of unusual importance. Quite aside from the significance and role in Nature of the cobalt to carbon bonds in the vitamin B 12 system and possible metal to carbon bonds in other biological systems, we need only consider that during the time taken to deliver this lec- ture, many thousands, if not tens of thousands of tons of chemical compounds are being transformed or synthesised industrially in processes which at some stage involve a transition metal to carbon bond. The nonchemist will pro- bably be most familiar with polyethylene or polypropylene in the form of do- mestic utensils, packaging materials, children’s toys and so on. These materials are made by Ziegler-Natta* or Philipps’ catalysis using titanium and chro- mium respectively. However, transition metal compounds are used as catalysts in the synthesis of synthetic rubbers and other polymers, and of a variety of simple compounds used as industrial solvents or intermediates. For example alcohols are made from olefins, carbon monoxide and hydrogen by use of cobalt or rhodium catalysts, acetic acid is made by carbonylation of methanol using rhodium catalysts and acrylonitrile is dimerised to adiponitrile (for nylon) by nickel catalysts. We should also not forget that the huge quantities of petroleum hydrocarbons processed by the oil and petrochemical industry are re-formed over platinum, platinum-rhenium or platinum-germanium sup- ported on alumina. -

Robert Burns Woodward
The Life and Achievements of Robert Burns Woodward Long Literature Seminar July 13, 2009 Erika A. Crane “The structure known, but not yet accessible by synthesis, is to the chemist what the unclimbed mountain, the uncharted sea, the untilled field, the unreached planet, are to other men. The achievement of the objective in itself cannot but thrill all chemists, who even before they know the details of the journey can apprehend from their own experience the joys and elations, the disappointments and false hopes, the obstacles overcome, the frustrations subdued, which they experienced who traversed a road to the goal. The unique challenge which chemical synthesis provides for the creative imagination and the skilled hand ensures that it will endure as long as men write books, paint pictures, and fashion things which are beautiful, or practical, or both.” “Art and Science in the Synthesis of Organic Compounds: Retrospect and Prospect,” in Pointers and Pathways in Research (Bombay:CIBA of India, 1963). Robert Burns Woodward • Graduated from MIT with his Ph.D. in chemistry at the age of 20 Woodward taught by example and captivated • A tenured professor at Harvard by the age of 29 the young... “Woodward largely taught principles and values. He showed us by • Published 196 papers before his death at age example and precept that if anything is worth 62 doing, it should be done intelligently, intensely • Received 24 honorary degrees and passionately.” • Received 26 medals & awards including the -Daniel Kemp National Medal of Science in 1964, the Nobel Prize in 1965, and he was one of the first recipients of the Arthur C. -

Los Premios Nobel De Química
Los premios Nobel de Química MATERIAL RECOPILADO POR: DULCE MARÍA DE ANDRÉS CABRERIZO Los premios Nobel de Química El campo de la Química que más premios ha recibido es el de la Quí- mica Orgánica. Frederick Sanger es el único laurea- do que ganó el premio en dos oca- siones, en 1958 y 1980. Otros dos también ganaron premios Nobel en otros campos: Marie Curie (física en El Premio Nobel de Química es entregado anual- 1903, química en 1911) y Linus Carl mente por la Academia Sueca a científicos que so- bresalen por sus contribuciones en el campo de la Pauling (química en 1954, paz en Física. 1962). Seis mujeres han ganado el Es uno de los cinco premios Nobel establecidos en premio: Marie Curie, Irène Joliot- el testamento de Alfred Nobel, en 1895, y que son dados a todos aquellos individuos que realizan Curie (1935), Dorothy Crowfoot Ho- contribuciones notables en la Química, la Física, la dgkin (1964), Ada Yonath (2009) y Literatura, la Paz y la Fisiología o Medicina. Emmanuelle Charpentier y Jennifer Según el testamento de Nobel, este reconocimien- to es administrado directamente por la Fundación Doudna (2020) Nobel y concedido por un comité conformado por Ha habido ocho años en los que no cinco miembros que son elegidos por la Real Aca- demia Sueca de las Ciencias. se entregó el premio Nobel de Quí- El primer Premio Nobel de Química fue otorgado mica, en algunas ocasiones por de- en 1901 al holandés Jacobus Henricus van't Hoff. clararse desierto y en otras por la Cada destinatario recibe una medalla, un diploma y situación de guerra mundial y el exi- un premio económico que ha variado a lo largo de los años. -

Cv Mlhg 2015
CURRICULUM VITAE Name: GREEN, Malcolm Leslie Hodder Address: St Catherine's College, Oxford or Inorganic Chemistry Laboratory South Parks Road OXFORD, OX1 3QR Date of Birth: 16/4/36, Eastleigh, Hampshire Nationality: British Marital Status: Married. Three children Degrees: B.Sc.(Hons), London; D.I.C., M.A.(Cantab), M.A.(Oxon), C.Chem., F.R.S.C., Ph.D., F.R.S. ACADEMIC CAREER 1953-56 Acton Technical College, University of London, B.Sc Hons. Chemistry 1956-59 Imperial College of Science and Technology, London; D.I.C. Ph.D. in chemistry. Supervisor Professor Sir G. Wilkinson 1959-60 Post-doctoral Research Associates Fellow. Imperial College of Science and Technology 1960-63 Assistant Lecturer in Inorganic Chemistry at Cambridge University 1961 Fellow of Corpus Christi College, Cambridge 1963 Sepcentenary Fellow of Inorganic Chemistry, Balliol College, Oxford and Departmental Demonstrator, University of Oxford 1965 University Lecturer, University of Oxford 1971 Visiting Professor, University of Western Ontario (Spring Term) 1972 Visiting Professor, Ecole de Chimie and Institute des Substances Naturelles, Paris (six months) 1973 A.P. Sloan Visiting Professor, Harvard University, (Spring Semester) 1979-84 Appointed to the British Gas Royal Society Senior Research Fellowship 1981 Sherman Fairchild Visiting Scholar at the California Institute of Technology(4 months) 1984 Re-appointed British Gas Royal Society Senior Research Fellow (1984-6) 1987 Vice-master, Balliol College, Oxford (T.T.) 1989 Appointed Professor of Inorganic Chemistry and Head of Department, Oxford University Fellow of St Catherine's College, Oxford 2004- present Emeritus Research Professor in the Inorganic Chemistry Laboratory, Oxford University Emeritus Fellow of Balliol College and St Catherine’s College Publications Two text books, 646 refereed papers and 8 patents. -

Guidelines and Suggested Title List for Undergraduate Chemistry Libraries, Serial Publication Number 44
DOCUMENT RESUME ED 040 037 SE 008 009 AUTHOR Marquardt, D. N., Ed. TITLE Guidelines and Suggested Title List for Undergraduate Chemistry Libraries, Serial Publication Number 44. INSTITUTION Advisory Council on Coll, Chemistry. PUB DATE Sep 69 NOTE 44p. AVAILABLE FROM Advisory Council on College Chemistry, Dept. of Chemistry, Stanford Univ., Stanford,California 94305 (free) EDRS PRICE EDRS P-: ice MF.40.25 HC-$2.30 DESCRIPTORS Advisory Committees, *Bibliographies,Booklists, *Chemistry, *College Science, *LibraryGuides, Research Reviews (Publications): *Resource Materials, Scholarly Journals IDENTIFIERS Advisory Council on College Chemistry ABSTRACT Contained are guidelines and an extensivelist of books and journals suitable for anundergraduate chemistry library. The guidelines are concerned with theorganization and acquisition policy of chemistry libraries, and withinter-library loan and photoduplication services. Various sections of the reportdeal with journals and abstracts, review serials,foreign language titles, U.S. Government publications and a suggestedtitles list. The books in the titles list are in the areas of analytical,biological, inorganic, organic and physical chemistry. Ingeneral, introductory texts have not been included. The list isarranged alphabetically with entries by author or editor unless the workis better known by title. The library of Congress classification numberand the Dewey Decimal classification number, when available, aregiven for each entry. Book prices are also given. The reportconcludes with a directory of publishers and dealers. This report shouldbe most useful for college libraries, science teachers, and students. (LC) 0 GUIDELINES AND SUGGESTEDTITLE LIST for t...UNDERGRADUATE CHEMISTRY LIBRARIES M CI Revised 1969 Co Co A Report Authorized by the ADVISORY COUNCIL ON COLLEGE CHEMISTRY Edited by D. -

Robert Burns Woodward 1917–1979
NATIONAL ACADEMY OF SCIENCES ROBERT BURNS WOODWARD 1917–1979 A Biographical Memoir by ELKAN BLOUT Any opinions expressed in this memoir are those of the author and do not necessarily reflect the views of the National Academy of Sciences. Biographical Memoirs, VOLUME 80 PUBLISHED 2001 BY THE NATIONAL ACADEMY PRESS WASHINGTON, D.C. ROBERT BURNS WOODWARD April 10, 1917–July 8, 1979 BY ELKAN BLOUT OBERT BURNS WOODWARD was the preeminent organic chemist Rof the twentieth century. This opinion is shared by his colleagues, students, and by other distinguished chemists. Bob Woodward was born in Boston, Massachusetts, and was an only child. His father died when Bob was less than two years old, and his mother had to work hard to support her son. His early education was in the Quincy, Massachusetts, public schools. During this period he was allowed to skip three years, thus enabling him to finish grammar and high schools in nine years. In 1933 at the age of 16, Bob Woodward enrolled in the Massachusetts Institute of Technology to study chemistry, although he also had interests at that time in mathematics, literature, and architecture. His unusual talents were soon apparent to the MIT faculty, and his needs for individual study and intensive effort were met and encouraged. Bob did not disappoint his MIT teachers. He received his B.S. degree in 1936 and completed his doctorate in the spring of 1937, at which time he was only 20 years of age. Immediately following his graduation Bob taught summer school at the University of Illinois, but then returned to Harvard’s Department of Chemistry to start a productive period with an assistantship under Professor E. -
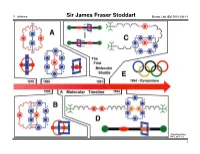
Sir James Fraser Stoddart Baran Lab GM 2010-08-14
Y. Ishihara Sir James Fraser Stoddart Baran Lab GM 2010-08-14 (The UCLA USJ, 2007, 20, 1–7.) 1 Y. Ishihara Sir James Fraser Stoddart Baran Lab GM 2010-08-14 (The UCLA USJ, 2007, 20, 1–7.) 2 Y. Ishihara Sir James Fraser Stoddart Baran Lab GM 2010-08-14 Professor Stoddart's publication list (also see his website for a 46-page publication list): - 9 textbooks and monographs - 13 patents "Chemistry is for people - 894 communications, papers and reviews (excluding book chapters, conference who like playing with Lego abstracts and work done before his independent career, the tally is about 770) and solving 3D puzzles […] - At age 68, he is still very active – 22 papers published in the year 2010, 8 months in! Work is just like playing - He has many publications in so many fields... with toys." - Journals with 10+ papers: JACS 75 Acta Crystallogr Sect C 26 ACIEE 67 JCSPT1 23 "There is a lot of room for ChemEurJ 62 EurJOC 19 creativity to be expressed JCSCC 51 ChemComm 15 in chemis try by someone TetLett 42 Carbohydr Res 12 who is bent on wanting to OrgLett 35 Pure and Appl Chem 11 be inventive and make JOC 28 discoveries." - High-profile general science journals: Nature 4 Science 5 PNAS 8 - Reviews: AccChemRes 8 ChemRev 4 ChemSocRev 6 - Uncommon venues of publication for British or American scientists: Coll. Czechoslovak Chem. Comm. 5 Mendeleev Communications 2 Israel Journal of Chemistry 5 Recueil des Trav. Chim. des Pays-Bas 2 Canadian Journal of Chemistry 4 Actualité chimique 1 Bibliography (also see his website, http://stoddart.northwestern.edu/ , for a 56-page CV): Chemistry – An Asian Journal 3 Bulletin of the Chem. -

Analysis of Nobel Prizes: the Place of Todmorden in the Annals of the Nobel Prize! As at the End of December 2017 Duncan Williamson
Analysis of Nobel Prizes: the place of Todmorden in the Annals of the Nobel Prize! as at the end of December 2017 Duncan Williamson Introduction My home town is Todmorden in West Yorkshire, England and throughout all of my childhood we were proud to say that Todmordian John Douglas Cockcroft had won the Nobel Prize for Physics in 1951: it was a joint award and he won it with Ernest Walton of Dungarvan, Ireland. We are told they were the first to have split the atom! In 1973 another Todmordian, Geoffrey Wilkinson won the Chemistry prize along with Ernst Otto Fischer for their work on sandwich compounds: they were working independently of one another, it seems. This put us in the stratosphere: which other town or city on the planet could boast TWO Nobel Prize Winners? Moreover, in spite of the 24 year age gap between them, Cockcroft and Wilkinson shared the same science teacher at Todmorden Grammar School. This article sets out to answer a series of questions I have never seen answered before which includes, is Todmorden unique in respect of it Nobel Prize achievements? Is Todmorden at the top of any Nobel list? Has any other town or city produced more than two Nobel Prizes. Has any town of the size of Todmorden or less produced two, or more, Nobel Prize winners? … all low level stuff but I could not find anywhere THE source that would tell me everything I wanted to know. Yes, the Official Web Site of the Nobel Prize, https://www.nobelprize.org, contains a massive amount of detail but it didn’t tell me, for example, if Todmorden is the smallest town to produce two Laureates and so on. -

Blue Hen Chemist #37 (August, 2010)
BLUE HEN CHEMIST University of Delaware, Department of Chemistry and Biochemistry Annual Alumni Newsletter NUMBER 37 AUGUST 2010 JOHN L. BURMEISTER, EDITOR Page ii August 2010 Blue Hen Chemist Number 37 Department of COVER: This is the 37th issue of the BLUE HEN CHEMIST and, accordingly, the cover picture shows a sample of natural alabaster (gypsum, CaS04 • 2H2O). Why? Because, as discovered by Jen Durkin, the 37th wedding anniversary is the alabaster anniversary! The distorted crys- tal structure of CaSO4 brings to mind the climactic battle scene in Avatar. —Sample courtesy of the University of Delaware Mineralogical Museum —Photo by Jen Durkin —Crystal structure provided by Prof. Svilen Bobev Blue Hen Chemist Number 37 August 2010 Page 1 CONTENTS From the Chair ...2 Professor John Hartwig to be Awarded 6th Annual Richard F. Heck Lectureship Award ...4 From the Associate Chair: ...5 Current Faculty URP Participants ...8 From the Director of Graduate Studies: ...9 Farewell to Joel Schneider ...12 Changes in the Wind for Introductory Chemistry ...13 Additional Faculty/Staff Activities and Awards: ...14 Visit us on Facebook! ...16 Postdoctoral Researchers and Fellows, 2009-10: ...17 Visiting Scholars, 2009-10: ...17 Let’s Communicate! ...17 Visiting Faculty, 2009 - 2010: ...18 2009-2010 Colloquia and Symposia: ...18 Graduate School Placements, 1994-2010 ...19 15th CHEM/BIOC Graduation Convocation, May 29, 2010 ...20 2009 - 2010 Undergraduate Awards ...22 2010 BA/BS/MA/MS/Ph.D. Graduates ...24 Fall 2010 Upcoming Seminars and Colloquia ...26 Graduate or Professional School Bound: ...27 Headed for Industry, Teaching, Etc.: ...27 2010 Graduate Student Placements: ...27 Alumni News ...28 Honor Roll of Gifts To The Department ...33 Kudos ...35 Giving To The Department ...36 Personal Information for CHEM/BIOC Records ...38 Page 2 August 2010 Blue Hen Chemist Number 37 FROM THE CHAIR Professor and Chair Klaus Theopold (b. -

Standard Lithium Welcomes Nobel Laureate, Professor Karl Barry Sharpless to Its Scientific Advisory Council
Standard Lithium Welcomes Nobel Laureate, Professor Karl Barry Sharpless to Its Scientific Advisory Council July 26, 2018 (Source) — Standard Lithium Ltd. (TSXV: SLL) (OTCQX: STLHF) (FRA: S5L) (“Standard Lithium” or the “Company”), is very pleased to announce the appointment of Nobel Laureate, Professor Barry Sharpless to the Company’s Scientific Advisory Council. Dr. Andy Robinson, President and COO of Standard Lithium commented, “It is truly a distinct honour to welcome Professor Sharpless, a chemist of global significance, to our Scientific Advisory Council.” Professor Sharpless is the W. M. Keck professor of chemistry at Scripps Research, where he has been a faculty member since 1990. He received the Nobel Prize in Chemistry in 2001 for his work on chirally catalyzed oxidation reactions. Since this landmark achievement, Prof. Sharpless has continued to be a luminary in the field, creating chemical tools that have been adopted by nearly every field of modern science. For his numerous contributions, the American Chemical Society (ACS) will award Professor Sharpless the 2019 Priestley Medal, the highest honor bestowed by ACS. His national and international awards include the inaugural Paul Janssen Prize for Creativity in Organic Synthesis, the King Faisal International Prize in Science, the Rhone Poulenc Medal, the Chemical Sciences Award of the U.S. National Academy of Sciences, the Benjamin Franklin Medal and the Wolf Prize. Standard Lithium CEO, Robert Mintak commented “I would like to thank Professors Jason Hein and Pierre Kennepohl of UBC for introducing Standard Lithium to Professor Sharpless. Knowing that the innovative and groundbreaking lithium processing work underway by Professors Hein, Kennepohl and our team is such that it piqued the interest of ProfessorSharpless is extremely rewarding, and we are thrilled to welcome Professor Sharpless to our Scientific Council“. -

Contributions of Civilizations to International Prizes
CONTRIBUTIONS OF CIVILIZATIONS TO INTERNATIONAL PRIZES Split of Nobel prizes and Fields medals by civilization : PHYSICS .......................................................................................................................................................................... 1 CHEMISTRY .................................................................................................................................................................... 2 PHYSIOLOGY / MEDECINE .............................................................................................................................................. 3 LITERATURE ................................................................................................................................................................... 4 ECONOMY ...................................................................................................................................................................... 5 MATHEMATICS (Fields) .................................................................................................................................................. 5 PHYSICS Occidental / Judeo-christian (198) Alekseï Abrikossov / Zhores Alferov / Hannes Alfvén / Eric Allin Cornell / Luis Walter Alvarez / Carl David Anderson / Philip Warren Anderson / EdWard Victor Appleton / ArthUr Ashkin / John Bardeen / Barry C. Barish / Nikolay Basov / Henri BecqUerel / Johannes Georg Bednorz / Hans Bethe / Gerd Binnig / Patrick Blackett / Felix Bloch / Nicolaas Bloembergen -

Exhibition and Sponsorship Prospectus
WLSC2016 世界生命科学大会 WORLD LIFE SCIENCE CONFERENCE 时间:2016年11月1日-3日 地点:中国.北京.国家会议中心 NOV.1-3, 2016 CHINA NATIONAL CONVENTION CENTER Exhibition and Sponsorship Prospectus Approver : The State Council, The People’s Republic of China Contact Information : Sponsor : China International Exchange Center for Science and Technology China Association for Science and Technology Address: 7 floor, No. 86 Xueyuan South Road, Haidian District, Beijing Organizer : Post/Zip Code: 100081 China Union of Life Science Societies Fax: 010-62380267 China International Exchange Center for Science and Technology Contact Name: Ms. Lu New Technology Development Center, China Association for Science and Technology E-mail: [email protected] MB: 13691041990 http://www.wlsc2016.com Contents Invitation The China Association of Science and Technology (CAST) and the China Union of Life Science Societies are organizing the 2016 World Life Science Conference on November 1-3, 2016 in Beijing, China. The conference is co-chaired by Prof. Qide Han, Vice Chairman of the National Committee of the Chinese People's Political Consultative Conference and President of CAST, and Prof. David Baltimore, the 1975 Nobel laureate in Physiology or Medicine. This conference will cover the latest and most exciting discoveries in basic research, technology development, government policies and science education in the areas of health, agriculture and Invitation 1 ………………………… environmental science. We have currently confirmed the attendance of 13 Nobel laureates, 3 World Food About Conference 2 … …………… Prize laureates, the President of the American Academy of Science, and the President of The Royal Society 4 General Information… ………… at the conference. Many leaders of international academic organizations and hundreds of outstanding About Exhibition… ……………10 scientists in the field of life sciences around the world will also be invited to attend.