Carbonyl Compounds Aldehydes and Ketones. on React of the Carbonyl Group
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
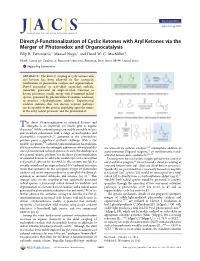
Direct Β‑Functionalization of Cyclic Ketones with Aryl Ketones Via the Merger of Photoredox and Organocatalysis Filip R
Communication pubs.acs.org/JACS Direct β‑Functionalization of Cyclic Ketones with Aryl Ketones via the Merger of Photoredox and Organocatalysis Filip R. Petronijevic,́† Manuel Nappi,† and David W. C. MacMillan* Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States *S Supporting Information ABSTRACT: The direct β-coupling of cyclic ketones with aryl ketones has been achieved via the synergistic combination of photoredox catalysis and organocatalysis. Diaryl oxymethyl or aryl−alkyl oxymethyl radicals, transiently generated via single-electron reduction of ketone precursors, readily merge with β-enaminyl radical species, generated by photon-induced enamine oxidation, to produce γ-hydroxyketone adducts. Experimental evidence indicates that two discrete reaction pathways can be operable in this process depending upon the nature of the ketyl radical precursor and the photocatalyst. he direct β-functionalization of saturated ketones and T aldehydes is an important yet elusive goal in organic chemistry.1 While carbonyl groups are readily amenable to ipso- and α-carbon substitution with a range of nucleophiles and electrophiles respectively,2,3 activation at the β-methylene position poses a significant synthetic challenge. With a few notable exceptions,1,4 carbonyl β-functionalization has tradition- ally been restricted to the conjugate addition of soft nucleophiles are accessed via carbene catalysis,9,10 nucleophilic addition of into α,β-unsaturated carbonyl systems. As such, the development acetal-protected Grignard reagents,11 or stoichiometric metal- 5 − of a general catalytic platform for the direct β-functionalization activated homoenolate equivalents.12 16 of saturated ketones or aldehydes would represent a conceptual Drawing from the mechanistic insights gained in the course of and practical advance for the field. -

Part I: Carbonyl-Olefin Metathesis of Norbornene
Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2016 1 © 2016 Zara Maxine Seibel All Rights Reserved 2 ABSTRACT Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel This thesis details progress towards the development of an organocatalytic carbonyl- olefin metathesis of norbornene. This transformation has not previously been done catalytically and has not been done in practical manner with stepwise or stoichiometric processes. Building on the previous work of the Lambert lab on the metathesis of cyclopropene and an aldehyde using a hydrazine catalyst, this work discusses efforts to expand to the less stained norbornene. Computational and experimental studies on the catalytic cycle are discussed, including detailed experimental work on how various factors affect the difficult cycloreversion step. The second portion of this thesis details the use of chiral cyclopropenimine bases as catalysts for asymmetric Michael reactions. The Lambert lab has previously developed chiral cyclopropenimine bases for glycine imine nucleophiles. The scope of these catalysts was expanded to include glycine imine derivatives in which the nitrogen atom was replaced with a carbon atom, and to include imines derived from other amino acids. i Table of Contents List of Abbreviations…………………………………………………………………………..iv Part I: Carbonyl-Olefin Metathesis…………………………………………………………… 1 Chapter 1 – Metathesis Reactions of Double Bonds………………………………………….. 1 Introduction………………………………………………………………………………. 1 Olefin Metathesis………………………………………………………………………… 2 Wittig Reaction…………………………………………………………………………... 6 Tebbe Olefination………………………………………………………………………... 9 Carbonyl-Olefin Metathesis……………………………………………………………. -

Catalytic Pyrolysis of Plastic Wastes for the Production of Liquid Fuels for Engines
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2019 Supporting information for: Catalytic pyrolysis of plastic wastes for the production of liquid fuels for engines Supattra Budsaereechaia, Andrew J. Huntb and Yuvarat Ngernyen*a aDepartment of Chemical Engineering, Faculty of Engineering, Khon Kaen University, Khon Kaen, 40002, Thailand. E-mail:[email protected] bMaterials Chemistry Research Center, Department of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen, 40002, Thailand Fig. S1 The process for pelletization of catalyst PS PS+bentonite PP ) t e PP+bentonite s f f o % ( LDPE e c n a t t LDPE+bentonite s i m s n HDPE a r T HDPE+bentonite Gasohol 91 Diesel 4000 3500 3000 2500 2000 1500 1000 500 Wavenumber (cm-1) Fig. S2 FTIR spectra of oil from pyrolysis of plastic waste type. Table S1 Compounds in oils (%Area) from the pyrolysis of plastic wastes as detected by GCMS analysis PS PP LDPE HDPE Gasohol 91 Diesel Compound NC C Compound NC C Compound NC C Compound NC C 1- 0 0.15 Pentane 1.13 1.29 n-Hexane 0.71 0.73 n-Hexane 0.65 0.64 Butane, 2- Octane : 0.32 Tetradecene methyl- : 2.60 Toluene 7.93 7.56 Cyclohexane 2.28 2.51 1-Hexene 1.05 1.10 1-Hexene 1.15 1.16 Pentane : 1.95 Nonane : 0.83 Ethylbenzen 15.07 11.29 Heptane, 4- 1.81 1.68 Heptane 1.26 1.35 Heptane 1.22 1.23 Butane, 2,2- Decane : 1.34 e methyl- dimethyl- : 0.47 1-Tridecene 0 0.14 2,2-Dimethyl- 0.63 0 1-Heptene 1.37 1.46 1-Heptene 1.32 1.35 Pentane, -

Aldehydes and Ketones
12 Aldehydes and Ketones Ethanol from alcoholic beverages is first metabolized to acetaldehyde before being broken down further in the body. The reactivity of the carbonyl group of acetaldehyde allows it to bind to proteins in the body, the products of which lead to tissue damage and organ disease. Inset: A model of acetaldehyde. (Novastock/ Stock Connection/Glow Images) KEY QUESTIONS 12.1 What Are Aldehydes and Ketones? 12.8 What Is Keto–Enol Tautomerism? 12.2 How Are Aldehydes and Ketones Named? 12.9 How Are Aldehydes and Ketones Oxidized? 12.3 What Are the Physical Properties of Aldehydes 12.10 How Are Aldehydes and Ketones Reduced? and Ketones? 12.4 What Is the Most Common Reaction Theme of HOW TO Aldehydes and Ketones? 12.1 How to Predict the Product of a Grignard Reaction 12.5 What Are Grignard Reagents, and How Do They 12.2 How to Determine the Reactants Used to React with Aldehydes and Ketones? Synthesize a Hemiacetal or Acetal 12.6 What Are Hemiacetals and Acetals? 12.7 How Do Aldehydes and Ketones React with CHEMICAL CONNECTIONS Ammonia and Amines? 12A A Green Synthesis of Adipic Acid IN THIS AND several of the following chapters, we study the physical and chemical properties of compounds containing the carbonyl group, C O. Because this group is the functional group of aldehydes, ketones, and carboxylic acids and their derivatives, it is one of the most important functional groups in organic chemistry and in the chemistry of biological systems. The chemical properties of the carbonyl group are straightforward, and an understanding of its characteristic reaction themes leads very quickly to an understanding of a wide variety of organic reactions. -

Aldehydes and Ketones Are Simple Organic Compounds Containing a Carbonyl Group
Aldehydes and Ketones are simple organic compounds containing a carbonyl group. Carbonyl group contains carbon- oxygen double bond. These organic compounds are simple because the carbon atom presents in the carbonyl group lack reactive groups such as OH or Cl. By Dr. Sayed Hasan Mehdi Assistant Professor Department of Chemistry Shia P.G. College, Lucknow Dr. S.Hasan Mehdi 6/13/2020 This is to bring to kind notice that the matter of this e- content is for the B.Sc. IV semester students. It has been taken from the following sources. The students are advised to follow these books as well. •A TEXTBOOK OF ORGANIC CHEMISTRY by Arun Bahl & B.S. Bahl, S. Chand & Company Ltd. Publication. •Graduate Organic Chemistry by M. K. Jain and S.C. Sharma, Vishal Publishing Co. •Pradeep’s Organic Chemistry Vol II by R. N. Dhawan, Pradeep Publication, Jalandhar. Dr. S.Hasan Mehdi 6/13/2020 An aldehyde is one of the classes of carbonyl group- containing alkyl group on one end and hydrogen on the other end. The R and Ar denote alkyl or aryl member respectively. In the condensed form, the aldehyde is written as –CHO. Dr. S.Hasan Mehdi 6/13/2020 Dr. S.Hasan Mehdi 6/13/2020 1. From Alcohols: a. By oxidation of Alcohols: Aldehydes and ketones can be prepared by the controlled oxidation of primary and secondary alcohols using an acidified solution of potassium dichromate or permanganate. Primary alcohol produces aldehydesRef. Last slide. O K Cr O RCH2OH + [O] 2 2 7 + R C H H 10 Alcohol Aldehyde O CH3CH2OH+ [O] K2Cr2O7 + CH3 C H H Ethyl Alcohol Acetaldehyde The aldehydes formed in the above reaction are very easily oxidised to carboxylc acids if allowed to remain in the reaction mixture. -

Aldehydes and Ketone
ALDEHYDES AND KETONE www.gneet.com ALDEHYDES AND KETONES In aldehydes, the carbonyl group is linked to either two hydrogen atom or one hydrogen atom and one carbon containing group such as alkyl, aryl or aralkyl group Examples * In ketones, the carbonyl group is linked to two carbon containing groups which may be same or different alkyl, aryl group. If two R and R’ groups are same, the ketone is called simple or symmetrical ketone and if R and R’ are different, then ketone is known as mixed or an unsymmetrical ketone. STRUCTURE Carbonyl carbon of both aldehyde and ketones is sp2 – hybridised, One of the three sp2 hybridised orbital get involved in σ- bond formation with half –filled p-orbital of oxygen atom whereas rest of the two are consumed in σ-bond formation with hydrogen and carbon depending on the structure of aldehyde or ketone. Unhybridised p-orbital of carbonyl carbon form π-bond with another half-filled p-orbital of oxygen atom by sideways overlapping. ISOMERISM IN ALDEHYDES AND KETONES (a) Chain isomerism: Aldehydes ( with 4 or more carbon atoms) and ketone ( with 5 or more carbon atoms) show chain isomerism. Example i) C4H8O CH3-CH2-CH2-CHO ( butanal) 1 ALDEHYDES AND KETONE www.gneet.com ii) C5H10O (b) Position isomerism: aliphatic aldehydes do not show position isomerism, because –CHO group is always present at the end of carbon chain. Aromatic aldehyde show position isomerism. Example (c) Metamerism: Higher ketones show metamerism due to presence of different alkyl groups attached to the same functional group C5H10O (d) Functional isomerism : Aldehydes and ketones show functional isomerism in them. -

Chapter 14 – Aldehydes and Ketones
Chapter 14 – Aldehydes and Ketones 14.1 Structures and Physical Properties of Aldehydes and Ketones Ketones and aldehydes are related in that they each possess a C=O (carbonyl) group. They differ in that the carbonyl carbon in ketones is bound to two carbon atoms (RCOR’), while that in aldehydes is bound to at least one hydrogen (H2CO and RCHO). Thus aldehydes always place the carbonyl group on a terminal (end) carbon, while the carbonyl group in ketones is always internal. Some common examples include (common name in parentheses): O O H HH methanal (formaldehyde) trans-3-phenyl-2-propenal (cinnamaldehyde) preservative oil of cinnamon O O propanone (acetone) 3-methylcyclopentadecanone (muscone) nail polish remover a component of one type of musk oil Simple aldehydes (e.g. formaldehyde) typically have an unpleasant, irritating odor. Aldehydes adjacent to a string of double bonds (e.g. 3-phenyl-2-propenal) frequently have pleasant odors. Other examples include the primary flavoring agents in oil of bitter almond (Ph- CHO) and vanilla (C6H3(OH)(OCH3)(CHO)). As your book says, simple ketones have distinctive odors (similar to acetone) that are typically not unpleasant in low doses. Like aldehydes, placing a collection of double bonds adjacent to a ketone carbonyl generally makes the substance more fragrant. The primary flavoring agent in oil of caraway is just a such a ketone. 2 O oil of carraway Because the C=O group is polar, small aldehydes and ketones enjoy significant water solubility. They are also quite soluble in typical organic solvents. 14.2 Naming Aldehydes and Ketones Aldehydes The IUPAC names for aldehydes are obtained by using rules similar to those we’ve seen for other functional groups (e.g. -

Carbonyl Compounds
CARBONYL COMPOUNDS PART-4, PPT-4, SEM-3 Dr. Kalyan Kumar Mandal Associate Professor St. Paul’s C. M. College Kolkata CONTENTS: CARBONYL COMPOUNDS PART-4 • Formation of Acetal/Ketal • Formation of Thioacetal Reaction of Carbonyl Compounds with Alcohols • Carbonyl compounds react with alcohols. The product of this reaction is known as a hemiacetal, because it is halfway to an acetal. This reaction is analogous to hydrate formation from aldehydes and ketones. The mechanism follows in the footsteps of hydrate formation: ROH is used instead of HOH (water). This Lecture is prepared by Dr. K. K. Mandal, SPCMC, Kolkata Formation of Cyclic Hemiacetal • Hemiacetal formation is reversible, and they are stabilized by the same special structural features as those of hydrates. However, hemiacetals can also gain stability by being cyclic. • Cyclic hemiacetal is formed when the carbonyl group and the attacking hydroxyl group are part of the same molecule. The reaction is an intramolecular (within the same molecule) addition, as opposed to the intermolecular (between two molecules) ones. This is an example of ring-chain tautomerism. This Lecture is prepared by Dr. K. K. Mandal, SPCMC, Kolkata Formation of Cyclic Hemiacetal • Although the cyclic hemiacetal is more stable, it is still in equilibrium with some of the open-chain hydroxyaldehyde form. Its stability, and how easily it forms, depend on the size of the ring. • Five- and six-membered rings involve less strain (their bonds are free to adopt 109° or 120° angles) in comparison to the three- membered rings, and therefore five or six-membered hemiacetals are very common. -

Conformational Studies of Aliphatic Secondary Ozonides (Propene, 1-Butene and 1-Heptene) by Means of FTIR Spectroscopy
DOI: 10.2478/s11532-006-0031-3 Research article CEJC 4(4) 2006 578–591 Conformational studies of aliphatic secondary ozonides (propene, 1-butene and 1-heptene) by means of FTIR spectroscopy Ruta Bariseviciute, Justinas Ceponkus, Alytis Gruodis, Valdas Sablinskas∗ Department of General Physics and Spectroscopy, Vilnius University, Vilnius-01513, Lithuania Received 6 January 2006; accepted 31 May 2006 Abstract: Ozonization reaction of simple alkenes was studied by means of FT infrared absorption gas spectroscopy. The reaction was performed at 95 K in neat films of the reactants. IR absorption spectra of the gaseous products were recorded. The spectra were analyzed combining experimental results with theoretical calculations performed at B3LYP 6-311++G (3df, 3pd) level. We found that among all theoretically predicted conformers of propene secondary ozonide, only one which has the O-O half-chair configuration for the five membered ring and the radical attached in the equatorial position was present in the sample. Samples of 1-butene and 1-heptene secondary ozonides consist from two conformers of very similar energy (ΔH =0.3 kJ/mol). The most stable conformer for both ozonides is the one with O-O half-chair configuration of the five membered ring and the radical attached in equatorial position and the aliphatic chain in gauche position. The second stable conformer has the aliphatic chain in anti position. c Versita Warsaw and Springer-Verlag Berlin Heidelberg. All rights reserved. Keywords: Ozonization, conformational analysis, secondary ozonides, IR spectroscopy 1 Introduction The ability of ozone to cleave double bonds in olefins has been known for more than 50 years. -

Aldehydes Can React with Alcohols to Form Hemiacetals
340 14 . Nucleophilic substitution at C=O with loss of carbonyl oxygen You have, in fact, already met some reactions in which the carbonyl oxygen atom can be lost, but you probably didn’t notice at the time. The equilibrium between an aldehyde or ketone and its hydrate (p. 000) is one such reaction. O HO OH H2O + R1 R2 R1 R2 When the hydrate reverts to starting materials, either of its two oxygen atoms must leave: one OPh came from the water and one from the carbonyl group, so 50% of the time the oxygen atom that belonged to the carbonyl group will be lost. Usually, this is of no consequence, but it can be useful. O For example, in 1968 some chemists studying the reactions that take place inside mass spectrometers needed to label the carbonyl oxygen atom of this ketone with the isotope 18 O. 16 18 By stirring the ‘normal’ O compound with a large excess of isotopically labelled water, H 2 O, for a few hours in the presence of a drop of acid they were able to make the required labelled com- í In Chapter 13 we saw this way of pound. Without the acid catalyst, the exchange is very slow. Acid catalysis speeds the reaction up by making a reaction go faster by raising making the carbonyl group more electrophilic so that equilibrium is reached more quickly. The the energy of the starting material. We 18 also saw that the position of an equilibrium is controlled by mass action— O is in large excess. -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

Pacs by Chemical Name (Mg/M3) (Pdf)
Table 4: Protective Action Criteria (PAC) Rev 25 based on applicable 60-minute AEGLs, ERPGs, or TEELs. Values are presented in mg/m3. August 2009 Table 4 is an alphabetical listing of the chemicals in the PAC data set. It provides Chemical Abstract Service Registry Numbers (CASRNs)1, PAC values, and technical information on the source of the PAC values. Table 4 presents all values for TEEL-0, PAC-1, PAC-2, and PAC-3 in mg/m3. The conversion of ppm to mg/m3 is calculated assuming 25 ºC and 760 mm Hg. The columns presented in Table 4 provide the following information: Heading Definition No. The ordered numbering of the chemicals as they appear in this alphabetical listing. Chemical Name The chemical name given to the PAC Development Team. CASRN The Chemical Abstract Service Registry Number for this chemical. TEEL-0 This is the threshold concentration below which most people will experience no adverse health effects. This PAC is always based on TEEL-0 because AEGL-0 or ERPG-0 values do not exist. PAC-1 Based on the applicable AEGL-1, ERPG-1, or TEEL-1 value. PAC-2 Based on the applicable AEGL-2, ERPG-2, or TEEL-2 value. PAC-3 Based on the applicable AEGL-3, ERPG-3, or TEEL-3 value. Source of PACs: Technical comments provided by the PAC development team that TEEL-0, PAC-1, indicate the source of the data used to derive PAC values. Future efforts PAC-2, PAC-3 are directed at reviewing, revising, and enhancing this information.