Conformational Studies of Aliphatic Secondary Ozonides (Propene, 1-Butene and 1-Heptene) by Means of FTIR Spectroscopy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Part I: Carbonyl-Olefin Metathesis of Norbornene
Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2016 1 © 2016 Zara Maxine Seibel All Rights Reserved 2 ABSTRACT Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel This thesis details progress towards the development of an organocatalytic carbonyl- olefin metathesis of norbornene. This transformation has not previously been done catalytically and has not been done in practical manner with stepwise or stoichiometric processes. Building on the previous work of the Lambert lab on the metathesis of cyclopropene and an aldehyde using a hydrazine catalyst, this work discusses efforts to expand to the less stained norbornene. Computational and experimental studies on the catalytic cycle are discussed, including detailed experimental work on how various factors affect the difficult cycloreversion step. The second portion of this thesis details the use of chiral cyclopropenimine bases as catalysts for asymmetric Michael reactions. The Lambert lab has previously developed chiral cyclopropenimine bases for glycine imine nucleophiles. The scope of these catalysts was expanded to include glycine imine derivatives in which the nitrogen atom was replaced with a carbon atom, and to include imines derived from other amino acids. i Table of Contents List of Abbreviations…………………………………………………………………………..iv Part I: Carbonyl-Olefin Metathesis…………………………………………………………… 1 Chapter 1 – Metathesis Reactions of Double Bonds………………………………………….. 1 Introduction………………………………………………………………………………. 1 Olefin Metathesis………………………………………………………………………… 2 Wittig Reaction…………………………………………………………………………... 6 Tebbe Olefination………………………………………………………………………... 9 Carbonyl-Olefin Metathesis……………………………………………………………. -

Catalytic Pyrolysis of Plastic Wastes for the Production of Liquid Fuels for Engines
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2019 Supporting information for: Catalytic pyrolysis of plastic wastes for the production of liquid fuels for engines Supattra Budsaereechaia, Andrew J. Huntb and Yuvarat Ngernyen*a aDepartment of Chemical Engineering, Faculty of Engineering, Khon Kaen University, Khon Kaen, 40002, Thailand. E-mail:[email protected] bMaterials Chemistry Research Center, Department of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen, 40002, Thailand Fig. S1 The process for pelletization of catalyst PS PS+bentonite PP ) t e PP+bentonite s f f o % ( LDPE e c n a t t LDPE+bentonite s i m s n HDPE a r T HDPE+bentonite Gasohol 91 Diesel 4000 3500 3000 2500 2000 1500 1000 500 Wavenumber (cm-1) Fig. S2 FTIR spectra of oil from pyrolysis of plastic waste type. Table S1 Compounds in oils (%Area) from the pyrolysis of plastic wastes as detected by GCMS analysis PS PP LDPE HDPE Gasohol 91 Diesel Compound NC C Compound NC C Compound NC C Compound NC C 1- 0 0.15 Pentane 1.13 1.29 n-Hexane 0.71 0.73 n-Hexane 0.65 0.64 Butane, 2- Octane : 0.32 Tetradecene methyl- : 2.60 Toluene 7.93 7.56 Cyclohexane 2.28 2.51 1-Hexene 1.05 1.10 1-Hexene 1.15 1.16 Pentane : 1.95 Nonane : 0.83 Ethylbenzen 15.07 11.29 Heptane, 4- 1.81 1.68 Heptane 1.26 1.35 Heptane 1.22 1.23 Butane, 2,2- Decane : 1.34 e methyl- dimethyl- : 0.47 1-Tridecene 0 0.14 2,2-Dimethyl- 0.63 0 1-Heptene 1.37 1.46 1-Heptene 1.32 1.35 Pentane, -

Properties and Synthesis. Elimination Reactions of Alkyl Halides
P1: PBU/OVY P2: PBU/OVY QC: PBU/OVY T1: PBU Printer: Bind Rite JWCL234-07 JWCL234-Solomons-v1 December 8, 2009 21:37 7 ALKENES AND ALKYNES I: PROPERTIES AND SYNTHESIS. ELIMINATION REACTIONS OF ALKYL HALIDES SOLUTIONS TO PROBLEMS 7.1 (a) ( E )-1-Bromo-1-chloro-1-pentene or ( E )-1-Bromo-1-chloropent-1-ene (b) ( E )-2-Bromo-1-chloro-1-iodo-1-butene or ( E )-2-Bromo-1-chloro-1-iodobut-1-ene (c) ( Z )-3,5-Dimethyl-2-hexene or ( Z )-3,5-Dimethylhex-2-ene (d) ( Z )-1-Chloro-1-iodo-2-methyl-1-butene or ( Z )-1-Chloro-1-iodo-2-methylbut-1-ene (e) ( Z,4 S )-3,4-Dimethyl-2-hexene or ( Z,4 S )-3,4-Dimethylhex-2-ene (f) ( Z,3 S )-1-Bromo-2-chloro-3-methyl-1-hexene or (Z,3 S )-1-Bromo-2-chloro-3-methylhex-1-ene 7.2 < < Order of increasing stability 7.3 (a), (b) H − 2 ∆ H° = − 119 kJ mol 1 Pt 2-Methyl-1-butene pressure (disubstituted) H2 − ∆ H° = − 127 kJ mol 1 Pt 3-Methyl-1-butene pressure (monosubstituted) H2 − ∆ H° = − 113 kJ mol 1 Pt 2-Methyl-2-butene pressure (trisubstituted) (c) Yes, because hydrogenation converts each alkene into the same product. 106 CONFIRMING PAGES P1: PBU/OVY P2: PBU/OVY QC: PBU/OVY T1: PBU Printer: Bind Rite JWCL234-07 JWCL234-Solomons-v1 December 8, 2009 21:37 ALKENES AND ALKYNES I: PROPERTIES AND SYNTHESIS 107 H H (d) > > H H H (trisubstituted) (disubstituted) (monosubstituted) Notice that this predicted order of stability is confirmed by the heats of hydro- genation. -
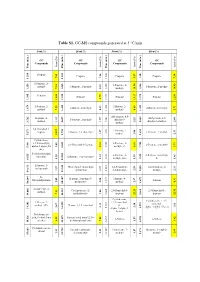
Table S1. GC-MS Compounds Generated at 5 °C/Min
Table S1. GC-MS compounds generated at 5 °C/min. 0 wt.% 10 wt.% 30 wt.% 50 wt.% GC GC GC GC Compounds Compounds Compounds Compounds Area (%) Area (%) Area (%) Area (%) Time (min.) (min.) Time Time (min.) (min.) Time (min.) Time (min.) Time Propene Propene Propene Propene 7.42 7.42 1.26 5.47 1.20 7.38 1.200 1.200 1.200 1.214 1.214 1-Propene, 2- 1-Propene, 2- methyl- 1-Propene, 2-methyl- 1-Propene, 2-methyl- 7.34 7.34 2.41 methyl- 1.245 1.245 1.245 1.259 1.259 10.54 1.246 14.90 Pentane Pentane Pentane Pentane 3.79 3.79 1.375 1.375 1.395 1.395 11.81 1.381 13.17 1.375 12.83 1-Pentene, 2- 2-Butene, 2- 2-Butene, 2-methyl- 2-Butene, 2-methyl- methyl- 7.25 0.93 methyl- 4.65 5.71 1.426 1.426 1.420 1.420 1.679 1.679 1H-Pyrazole, 4,5- Heptane, 4- 1H-Pyrazole, 4,5- 1-Pentene, 2-methyl- dihydro-5- methyl- 1.92 1.57 2.25 dihydro-5-methyl- 2.44 1.659 1.659 1.562 1.569 3.762 3.762 methyl- 2,4-Dimethyl-1- 1-Pentene, 2- heptene 2-Butene, 2,3-dimethyl- 1-Pentene, 2-methyl- 1.27 1.27 methyl- 3.08 2.33 1.653 1.653 1.653 5.671 5.671 34.57 1.750 Cyclohexane, 1,3,5-trimethyl-, 2-Pentene, 3- 2,4-Dimethyl-1-heptene 2-Pentene, 2-methyl- alpha.,3.alpha.,5.b 2.03 9.11 methyl-, €- 6.11 5.73 5.605 5.605 1.743 1.744 5.981 5.981 eta.)- 5-Aminoisoxazole 2-Pentene, 3- 2-Pentene, 3-methyl-, Spermine Ethanone, 1-cyclopentyl- 1.55 1.55 1.32 methyl-, (Z)- 1.59 €- 1.46 1.828 1.828 1.828 6.925 6.925 10.560 Ethanone, 1- 1R,2c,3t,4t-Tetramethyl- 1,3-Pentadiene, 1,4-Hexadiene, 5- cyclopentyl- 3.57 3.57 cyclohexan 0.97 2,3-dimethyl- 2.28 methyl- 2.31 2.584 2.584 2.585 10.613 10.613 10.638 N- Heptane, 2-methyl-3- Heptane, 4- Methylallylamine Toluene 2.49 2.49 methylene- 2.50 methyl- 2.97 4.31 3.697 3.697 3.697 14.047 14.047 10.684 10.684 2-Undecene, 4- Cyclopentane, (2- 2,4-Dimethyl-1- 2,4-Dimethyl-1- methyl- 7.81 7.81 methylbutyl)- 1.00 heptene heptene 5.605 5.605 22.29 22.29 5.605 12.95 14.086 14.086 14.163 Cyclohexane, Cyclohexane, 1,3,5- 2-Decene, 7- 1,3,5-trimethyl-, trimethyl- methyl-, (Z)- Hexane, 2,3,4-trimethyl- (1. -

United States Patent Office
Patented July 16, 1957 United States Patent Office 1. 2 be isolated from a solution in petroleum ether by frac tional crystallization. 2,799,685 The homologues, i. e. for example the pentachloro and tetrachloro-derivatives and the corresponding ana UNSATURATEDPROCESS FOR POLYCYCLIC THEIR MANUFACTURE SULFITES AND A logues of methyl and ethyl can be obtained in a corre einz Frensch, Helmut Goebel, Wilhelm Staudermann, sponding manner. : and Walter Finkenbrink, Frankfurt am Main, Ger. As Diels-Alder-components suitable for the manufac many, assignors to Farbwerke Hoechst Aktiengesell ture of Sulfites of the kind described above, there come schaft yormals Meister Lucius & Brining, Frankfurt also into consideration the halogenated alkyl derivatives, am Main, Germany; a corporation of the Federal Re O for example, dimethylcyclopentadiene. public of Germany The polycyclic halogenated sulfites are readily crystal lizing colorless compounds. No Drawing. Application April 8, 1955, The products of this invention may be used for pro Serial No. 500,264 tecting wood, paper, textiles and leather and as inter Claims priority, application Germany April 17, 1954 5 mediates for the manufacture of drugs. In the pure state, they are practically, odourless and are, therefore, 9 Claims. (CI. 260-327) especially suitable for the mentioned purposes. They The present invention provides new unsaturated poly have a curative and prophylactic effect and are prefer cyclic sulfites corresponding to the following general ably used, if desired in admixture with other insecticides, 20 ovicides, fungicides, herbicides or fertilizers, for combat formula: ing animal and fungoid pests. Y - In addition to their excellent action as pesticides the substances hereinbefore described are distinguished by 4R/V their chemical stability. -

Gfsorganics & Fragrances
Chemicals for Flavors GFSOrganics & Fragrances GFS offers a broad range of specialty organic chemicals Specialized chemistries we as building blocks and intermediates for the manufacture of offer include: flavors and fragrances. • Alkynes Over 5,500 materials, including 1,400 chemicals from natural sources, are used for flavor • Alkynols enhancements and aroma profiles. These aroma chemicals are integral components of • Olefins the continued growth within consumer products and packaged foods. The diversity of products can be attributed to the broad spectrum of organic compounds derived from • Unsaturated Acids esters, aldehydes, lactones, alcohols and several other functional groups. • Unsaturated Esters • DIPPN and other Products GFS Chemicals manufactures a wide range of organic intermediates that have been utilized in a multitude of personal care applications. For example, we support Why GFS? several market leading companies in the manufacture and supply of alkyne based aroma chemicals. • Specialized Chemistries • Tailored Specifications As such, we understand the demanding nature of this fast changing market and are fully • From Grams to Metric Tons equipped to overcome process challenges and manufacture the novel chemical products • Responsive Technical Staff that you need, when you need them. We offer flexible custom manufacturing services to produce high purity products with the assurance of quality and full confidentiality. • Uncompromised Product Quality Our state-of-the-art manufacturing facility, located in Columbus, OH is ISO 9001:2008 certified. As a U.S. based manufacturer we have a proven record of helping you take products from development to commercialization. Our technical sales experts are readily accessible to discuss your project needs and unique product specifications. -

Elimination Reactions Are Described
Introduction In this module, different types of elimination reactions are described. From a practical standpoint, elimination reactions widely used for the generation of double and triple bonds in compounds from a saturated precursor molecule. The presence of a good leaving group is a prerequisite in most elimination reactions. Traditional classification of elimination reactions, in terms of the molecularity of the reaction is employed. How the changes in the nature of the substrate as well as reaction conditions affect the mechanism of elimination are subsequently discussed. The stereochemical requirements for elimination in a given substrate and its consequence in the product stereochemistry is emphasized. ELIMINATION REACTIONS Objective and Outline beta-eliminations E1, E2 and E1cB mechanisms Stereochemical considerations of these reactions Examples of E1, E2 and E1cB reactions Alpha eliminations and generation of carbene I. Basics Elimination reactions involve the loss of fragments or groups from a molecule to generate multiple bonds. A generalized equation is shown below for 1,2-elimination wherein the X and Y from two adjacent carbon atoms are removed, elimination C C C C -XY X Y Three major types of elimination reactions are: α-elimination: two atoms or groups are removed from the same atom. It is also known as 1,1-elimination. H R R C X C + HX R Both H and X are removed from carbon atom here R Carbene β-elimination: loss of atoms or groups on adjacent atoms. It is also H H known as 1,2- elimination. R C C R R HC CH R X H γ-elimination: loss of atoms or groups from the 1st and 3rd positions as shown below. -

Nuclear Spin-Induced Optical Rotation of Functional Groups in Hydrocarbons Petr Štěpánek1 Supplementary Information
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is © the Owner Societies 2020 Nuclear spin-induced optical rotation of functional groups in hydrocarbons Petr Štěpánek1 Supplementary Information Contents 1 List of molecules 2 2 Basis set benchmark 5 1 3 Effect of cis/trans isomerism on H NSOR of T1 atom type in alkenes 5 4 Effect of conformation on NSOR 6 5 Extended Figure for NSOR of dienes 7 6 Effect of cis/trans isomerism in dienes 8 7 Supporting data for solvent and geometry effects 8 1NMR Research Unit, Faculty of Science, University of Oulu, P.O. Box 3000, FI-90014 Oulu, Finland; petr.stepanek@oulu.fi 1 1 List of molecules Tables below list the molecules included in the study. The test cases of the contribution model are not included. Table S 1: Alkane molecules methane 2-methyl-propane 2,3-dimethyl-butane ethane 2-methyl-butane 2,2-dimethyl-propane propane 2-methyl-pentane 2,2-dimethyl-pentane butane 3-methyl-pentane 2,3-dimethyl-pentane pentane 2,2-dimethyl-butane 3,3-dimethyl-pentane hexane Table S 2: Alkene molecules ethene 2,3,3-trimethyl-butene 5-methyl-heptene propene 2,4-dimethyl-pentene 6-methyl-heptene trans-but-2-ene 2-methyl-hexene cis-5,5-dimethyl-hex-2-ene cis-pent-2-ene 3,3-dimethyl-pentene cis-6-methyl-hept-2-ene trans-hex-2-ene 3,4-dimethyl-pentene trans-5,5-dimethyl-hex-2-ene trans-pent-2-ene 3-methyl-hexene trans-6-methyl-hept-2-ene trans-hex-3-ene 4-methyl-hexene 5,6-dimethyl-heptene 2-methyl-propene cis-4,4-dimethyl-pent-2-ene cis-6,6-dimethyl-hepte-2-ene 2-methyl-butene -

Carbene Rearrangements: Intramolecular Interaction of a Triple Bond with a Carbene Center
An Abstract OF THE THESIS OF Jose C. Danino for the degree of Doctor of Philosophy in Chemistry presented on _Dcc, Title: Carbene RearrangementE) Intramolecular Interaction of a Triple Bond with aCarbene Center Redacted for Privacy Abstract approved: Dr. Vetere. Freeman The tosylhydrazones of2-heptanone, 4,4-dimethy1-2- heptanone, 6-heptyn-2-one and 4,4-dimethy1-6-heptyn-2- one were synthesizedand decomposed under a varietyof reaction conditions:' drylithium and sodium salt pyrolyses, sodium methoxide thermolysesin diglyme and photolyses of the lithium salt intetrahydrofuran. The saturated ana- logues 2-heptanone tosylhydrazoneand its 4,4-dimethyl isomer afforded the alkenesarising from 6-hydrogeninser- product distribution in the tion. It was determined that differ- dry salt pyrolyses of2-heptanone tosylhydrazone was ent for the lithiumand the sodium salts. However, the product distribution of thedry sodium salt was verysimilar diglyme to product distributionobtained on thermolysis in explained by a with sodium methoxide. This difference was reaction of lithium bromide(present as an impurity inall compound to the lithium salts)with the intermediate diazo afford an organolithiumintermediate that behaves in a some- what different fashionthan the free carbene.The unsaturated analogues were found to produce a cyclic product in addition to the expected acyclic alkenes arising from 3-hydrogen insertion. By comparison of the acyclic alkene distri- bution obtained in the saturated analogues with those in the unsaturated analogues, it was concluded that at leastsome cyclization was occurring via addition of the diazo moiety to the triple bond. It was determined that the organo- lithium intermediateresulting from lithium bromide cat- alyzed decomposition of the diazo compound was incapable of cyclization. -

Chapter 21 Organic Chemistry
CHAPTER 21 ORGANIC CHEMISTRY Hydrocarbons 1. A hydrocarbon is a compound composed of only carbon and hydrogen. A saturated hydro- carbon has only carbon-carbon single bonds in the molecule. An unsaturated hydrocarbon has one or more carbon-carbon multiple bonds but may also contain carbon-carbon single bonds. A normal hydrocarbon has one chain of consecutively bonded carbon atoms. A branched hydrocarbon has at least one carbon atom not bonded to the end carbon of a chain of consecutively bonded carbon atoms. Instead, at least one carbon atom forms a bond to an inner carbon atom in the chain of consecutively bonded carbon atoms. 2. To determine the number of hydrogens bonded to the carbons in cyclic alkanes (or any alkane where they may have been omitted), just remember that each carbon has four bonds. In cycloalkanes, only the C−C bonds are shown. It is assumed you know that the remaining bonds on each carbon are C−H bonds. The number of C−H bonds is that number required to give the carbon four total bonds. 3. In order to form, cyclopropane and cyclobutane are forced to form bond angles much smaller than the preferred 109.5° bond angles. Cyclopropane and cyclobutane easily react in order to obtain the preferred 109.5° bond angles. 4. Aromatic hydrocarbons are a special class of unsaturated hydrocarbons based on the benzene ring. Benzene has the formula C6H6. It is a planar molecule (all atoms are in the same plane). Each carbon in benzene is attached to three other atoms; it exhibits trigonal planar geometry with 120° bond angles. -

United States Patent Office
2,841,485 United States Patent Office Patented July 1, 1958 2 1,4,5,6,7,7-hexachloro - 2 - methylbicyclo[2.2.11 - 5 2,841,485 heptene-2,3-diol cyclic carbonate, HEXAHALOBICYCLOESEPTENEDOLS AND 1,4,5,6,7,7-hexachioro 2 "ethylbicyclo[2.2.11 - 5 DERVATIVES THEREOF heptene-2,3-diol cyclic carbonate, 1,4,5,6,7,7 - hexachloro - 2 - phenylbicyclo[2.2.1 - 5 William K. Johnson, Dayton, Ohio, and Tad Le Marre heptene-2,3-diol cyclic carbonate, Patton, Houston, Tex., assignors to Monsanto Chemi 1,4,5,6,7,7-hexachloro - 2,3-dimethylbicyclo[2.2.1 - 5 cal Company, St. Louis, Mo., a corporatio of Dela heptene-2,3-diol cyclic carbonate, ware 1,4,5,6,7,7-hexachloro - 2 - methyl - 3 - phenylbi No Drawing. Application May 20, 1955 0. cyclo2.2.1]-5-heptene-2,3-diol cyclic carbonate, Serial No. 510,053 1,4,5,6,7,7-hexabromobicyclo[2.2.1] - 5 - heptene - 2,3- diol cyclic carbonate, 26 Claims. (C. 71-23) 1,4,5,6 - tetrachloro - 7,7 - difluorobicyclo[2.2.1 - 5 heptene-2,3-diol cyclic carbonate, etc. This invention relates to hexahalobicycloheptenediols and derivatives thereof, to methods of making the same, The preparation of 1,4,5,6,7,7 - hexachlorobicyclo and to the use of these compounds as biological (2.2.1]-5-heptene-2,3-diol carbonate, for example, may toxicants. - be carried out as follows: The present invention provides new and useful com Example 1 pounds of the general formula. -

Processes for Separation of Fluoroolefins From
(19) TZZ Z¥__T (11) EP 2 054 361 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C07C 17/38 (2006.01) C07C 17/383 (2006.01) 17.02.2016 Bulletin 2016/07 C07C 17/386 (2006.01) C01B 7/19 (2006.01) C07C 17/25 (2006.01) (21) Application number: 07811541.7 (86) International application number: (22) Date of filing: 24.08.2007 PCT/US2007/018837 (87) International publication number: WO 2008/024508 (28.02.2008 Gazette 2008/09) (54) PROCESSES FOR SEPARATION OF FLUOROOLEFINS FROM HYDROGEN FLUORIDE BY AZEOTROPIC DISTILLATION VERFAHREN ZUR TRENNUNG VON FLUOROLEFINEN VON WASSERSTOFFFLUORID DURCH AZEOTROPE DESTILLATION PROCÉDÉS DE SÉPARATION DE FLUOROOLÉFINES DU FLUORURE D’HYDROGÈNE PAR DISTILLATION AZÉOTROPIQUE (84) Designated Contracting States: • MAHLER, Barry, Asher DE ES FR GB IT PL Glen Mills, Pennsylvania 19342 (US) • TOTON, Donald, J. (30) Priority: 24.08.2006 US 839737 P New Castle, Delaware 19720 (US) (43) Date of publication of application: (74) Representative: Matthews, Derek Peter et al 06.05.2009 Bulletin 2009/19 Dehns St Bride’s House (60) Divisional application: 10 Salisbury Square 11174594.9 London EC4Y 8JD (GB) 11174764.8 / 2 433 921 (56) References cited: (73) Proprietor: E. I. du Pont de Nemours and Company WO-A-99/20585 WO-A-99/26907 Wilmington, DE 19898 (US) US-A1- 2006 116 538 US-B1- 6 303 838 (72) Inventors: Remarks: • KNAPP, Jeffrey, P. Thefile contains technical information submitted after Wilmington, Delaware 19808 (US) the application was filed and not included in this specification Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations.