Novel Mutation of Type 1 Insulin-Like Growth Factor Receptor (IGF1R) Gene in a Severe Short Stature Pedigree Identified by Targeted Next-Generation Sequencing
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

TTDN1 (MPLKIP) (NM 138701) Human 3' UTR Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for SC204348 TTDN1 (MPLKIP) (NM_138701) Human 3' UTR Clone Product data: Product Type: 3' UTR Clones Product Name: TTDN1 (MPLKIP) (NM_138701) Human 3' UTR Clone Vector: pMirTarget (PS100062) Symbol: MPLKIP Synonyms: ABHS; C7orf11; ORF20; TTD4 ACCN: NM_138701 Insert Size: 2000 bp This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 3 TTDN1 (MPLKIP) (NM_138701) Human 3' UTR Clone – SC204348 Insert Sequence: >SC204348 3’UTR clone of NM_138701 The sequence shown below is from the reference sequence of NM_138701. The complete sequence of this clone may contain minor differences, such as SNPs. Blue=Stop Codon Red=Cloning site GGCAAGTTGGACGCCCGCAAGATCCGCGAGATTCTCATTAAGGCCAAGAAGGGCGGAAAGATCGCCGTG TAACAATTGGCAGAGCTCAGAATTCAAGCGATCGCC ACAGGCAAAAAAGGAAGATACTTTTGTTAACATTTCTGAAATTCAACTGGAAGCTTCATGTGTCAGGAA CATCTTGGACAAAACTTTAAGTTGTGTTGATATAAATTTACCCAAAGATGATGACTTTGATTGGATAAT TAGTAAGGTCTTTTTGTTATTTTTCATCGTATCAGGTATTGTTGATATTAGAGAAAAAAGTAGGATAAC TTGCAACATTTAGCTCTGGAAGTACCTACCACATTTTAGAGATTTACCGTTTCCATATATTTAACATTC CTGGTTACATAATGGACATTTGTCTTTTAATGTTTTTTCAATGTTTTAAAATAAAACATTTTGTCTTCT AGCTATTGTGGTTTTGTGGTATGATAAAGAAGTAGACTTACTACAGTAATGCTTTGTAGTCACTTAGAG TTCATAGGTAAATGTTTTGCAAATTATTTTTGAAAATGAAATAGGTAAACCATCCTTTGAGCTGTAGAC -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Bioinformatics Analysis for the Identification of Differentially Expressed Genes and Related Signaling Pathways in H
Bioinformatics analysis for the identification of differentially expressed genes and related signaling pathways in H. pylori-CagA transfected gastric cancer cells Dingyu Chen*, Chao Li, Yan Zhao, Jianjiang Zhou, Qinrong Wang and Yuan Xie* Key Laboratory of Endemic and Ethnic Diseases , Ministry of Education, Guizhou Medical University, Guiyang, China * These authors contributed equally to this work. ABSTRACT Aim. Helicobacter pylori cytotoxin-associated protein A (CagA) is an important vir- ulence factor known to induce gastric cancer development. However, the cause and the underlying molecular events of CagA induction remain unclear. Here, we applied integrated bioinformatics to identify the key genes involved in the process of CagA- induced gastric epithelial cell inflammation and can ceration to comprehend the potential molecular mechanisms involved. Materials and Methods. AGS cells were transected with pcDNA3.1 and pcDNA3.1::CagA for 24 h. The transfected cells were subjected to transcriptome sequencing to obtain the expressed genes. Differentially expressed genes (DEG) with adjusted P value < 0.05, | logFC |> 2 were screened, and the R package was applied for gene ontology (GO) enrichment and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. The differential gene protein–protein interaction (PPI) network was constructed using the STRING Cytoscape application, which conducted visual analysis to create the key function networks and identify the key genes. Next, the Submitted 20 August 2020 Kaplan–Meier plotter survival analysis tool was employed to analyze the survival of the Accepted 11 March 2021 key genes derived from the PPI network. Further analysis of the key gene expressions Published 15 April 2021 in gastric cancer and normal tissues were performed based on The Cancer Genome Corresponding author Atlas (TCGA) database and RT-qPCR verification. -

MPLKIP Gene M-Phase Specific PLK1 Interacting Protein
MPLKIP gene M-phase specific PLK1 interacting protein Normal Function The MPLKIP gene (formerly known as C7orf11) provides instructions for making a protein called M-phase specific PLK1 interacting protein. The function of this protein is unclear. Based on its interaction with a protein called Plk1, the MPLKIP protein is thought to play a role in cell growth and division. In particular, it may help regulate the cell cycle, which is the cell's way of replicating itself in an organized, step-by-step fashion. Researchers speculate that the MPLKIP protein may also be involved in gene transcription, which is the first step in protein production. Health Conditions Related to Genetic Changes Trichothiodystrophy At least eight mutations in the MPLKIP gene have been identified in people with trichothiodystrophy. These mutations cause some cases of the non-photosensitive form of the disorder, which is not associated with extreme sensitivity to ultraviolet (UV) rays from sunlight. All of the known MPLKIP gene mutations prevent the production of any functional MPLKIP protein. It is unknown how a loss of this protein leads to the characteristic features of trichothiodystrophy, including slow growth, intellectual disability, and brittle hair. Other Names for This Gene • ABHS • C7orf11 • chromosome 7 open reading frame 11 • ORF20 • TTD non-photosensitive 1 protein • TTDN1 • TTDN1_HUMAN Reprinted from MedlinePlus Genetics (https://medlineplus.gov/genetics/) 1 A dditional Information & Resources Tests Listed in the Genetic Testing Registry • Tests of MPLKIP -

Original Article Identification of Differentially Expressed Genes Between Male and Female Patients with Acute Myocardial Infarction Based on Microarray Data
Int J Clin Exp Med 2019;12(3):2456-2467 www.ijcem.com /ISSN:1940-5901/IJCEM0080626 Original Article Identification of differentially expressed genes between male and female patients with acute myocardial infarction based on microarray data Huaqiang Zhou1,2*, Kaibin Yang2*, Shaowei Gao1, Yuanzhe Zhang2, Xiaoyue Wei2, Zeting Qiu1, Si Li2, Qinchang Chen2, Yiyan Song2, Wulin Tan1#, Zhongxing Wang1# 1Department of Anesthesiology, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou, China; 2Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, China. *Equal contributors and co-first au- thors. #Equal contributors. Received May 31, 2018; Accepted August 4, 2018; Epub March 15, 2019; Published March 30, 2019 Abstract: Background: Coronary artery disease has been the most common cause of death and the prognosis still needs further improving. Differences in the incidence and prognosis of male and female patients with coronary artery disease have been observed. We constructed this study hoping to understand those differences at the level of gene expression and to help establish gender-specific therapies. Methods: We downloaded the series matrix file of GSE34198 from the Gene Expression Omnibus database and identified differentially expressed genes between male and female patients. Gene ontology, Kyoto Encyclopedia of Genes and Genomes pathway enrichment analy- sis, and GSEA analysis of differentially expressed genes were performed. The protein-protein interaction network was constructed of the differentially expressed genes and the hub genes were identified. Results: A total of 215 up-regulated genes and 353 down-regulated genes were identified. The differentially expressed pathways were mainly related to the function of ribosomes, virus, and related immune response as well as the cell growth and proliferation. -

Molecular Mechanisms and the Conflict Between
Molecular Ecology (2016) 25, 4368–4376 doi: 10.1111/mec.13766 Molecular mechanisms and the conflict between courtship and aggression in three-spined sticklebacks YIBAYIRI O. SANOGO and ALISON M. BELL School of Integrative Biology, Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana-Champaign, 505 S. Goodwin Ave., 433 Morrill Hall, Urbana, IL 61801, USA Abstract In nature, animals often face conflicting demands. For example, breeding males must attract a mate but at the same time be ready to defend against rivals. The molecular mechanisms by which the brain resolves behavioural trade-offs are largely unknown. In this study, we compared the brain transcriptional responses of territorial male three-spined sticklebacks to a mating opportunity with a female and to a territorial challenge by a rival male. We focused on the diencephalon and the cerebellum, two regions of the brain implicated in courtship and aggression. There was a set of genes that were differentially expressed in response to both a courtship opportunity and a territorial challenge. Closer inspection of the direction of regulation revealed that genes that were downregulated in response to a courtship opportunity were upregu- lated in response to a territorial challenge and vice versa. Our study reveals some of the potential molecular mechanisms underlying behavioural trade-offs between sex and aggression, along with a possible solution to the conflict via social context- dependent gene regulation. Keywords: behavioural syndrome, Gasterosteus aculeatus, gene expression, limited plasticity, microarray, sociogenomics, territoriality Received 8 April 2015; revision received 28 June 2016; accepted 19 July 2016 the molecular mechanisms by which the brain might Introduction generate and/or resolve such conflicts is in its infancy Many animals in nature are constantly confronted with (O’Connell & Hofmann 2011a). -

Screening of Exosomal Mirnas Derived From
3314 MOLECULAR MEDICINE REPORTS 18: 3314-3324, 2018 Screening of exosomal miRNAs derived from subcutaneous and visceral adipose tissues: Determination of targets for the treatment of obesity and associated metabolic disorders ZHENG YANG1*, ZHUYING WEI1*, XIA WU2 and HUIDI YANG1 1Basic Medical School, Inner Mongolia Medical University, Hohhot, Inner Mongolia 010110; 2The State Key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock, Inner Mongolia University, Hohhot, Inner Mongolia 010070, P.R. China Received November 1, 2017; Accepted June 14, 2018 DOI: 10.3892/mmr.2018.9312 Abstract. Exosomal micro (mi)RNAs have been suggested 58 DE‑miRNAs were identified in VAT between patients with to have important roles in abdominal obesity, and to be obesity and lean patients. miRNA (miR)-4517 was revealed to associated with metabolic alterations via posttranscriptional be a downregulated exosomal miRNA between SAT and VAT, regulation of target genes. However, exosomal miRNA while the other DE-miRNAs were SAT-(e.g. hsa-miR-3156-5p profiles in subcutaneous adipose tissue (SAT) and visceral and hsa-miR-4460) or VAT-(e.g. hsa-miR-582-5p, adipose tissue (VAT) have rarely been investigated. In the hsa‑miR‑566 and miR‑548) specific. Following overlapping present study, microarray data were obtained from the Gene with the target genes of DE-miRNAs, only one DEG [cluster Expression Omnibus database with the following accession of differentiation 86 (CD86)] was identified in SAT samples, numbers: GSE68885 (exosomal miRNAs in SAT obtained whereas 25 DEGs (e.g. fibroblast growth factor 2 (FGF2), from seven patients with obesity and five lean patients), FOS like 2, AP‑1 transcription factor subunit (FOSL2); and GSE50574 (exosomal miRNAs in VAT obtained from seven adenosine monophosphate deaminase 3 (AMPD3)] were iden- patients with obesity and five lean patients) and GSE29718 tified in VAT samples. -
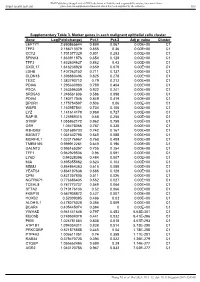
Value Cluster LEFTY1 2.690865644 0.899 0.067 0.00E+00 C1
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut Supplementary Table 3. Marker genes in each malignant epithelial cells cluster Gene Log(Fold-change) Pct.1 Pct.2 Adj p- value Cluster LEFTY1 2.690865644 0.899 0.067 0.00E+00 C1 TFF2 2.186713379 0.855 0.36 0.00E+00 C1 CCT2 1.701377329 0.801 0.283 0.00E+00 C1 SPINK4 1.663911876 0.654 0.128 0.00E+00 C1 TFF3 1.632604627 0.882 0.43 0.00E+00 C1 CXCL17 1.616238928 0.659 0.079 0.00E+00 C1 LDHB 1.407263152 0.711 0.137 0.00E+00 C1 CLDN18 1.398880496 0.825 0.278 0.00E+00 C1 TESC 1.382790712 0.79 0.212 0.00E+00 C1 PDIA6 1.295630983 0.789 0.404 0.00E+00 C1 PSCA 1.263596359 0.922 0.241 0.00E+00 C1 SRD5A3 1.246561606 0.586 0.098 0.00E+00 C1 PDIA4 1.180717046 0.849 0.419 0.00E+00 C1 DPCR1 1.175754587 0.506 0.06 0.00E+00 C1 WBP5 1.163957541 0.734 0.106 0.00E+00 C1 LYZ 1.141614179 0.969 0.727 0.00E+00 C1 RAP1B 1.125989315 0.68 0.255 0.00E+00 C1 S100P 1.055862172 0.962 0.758 0.00E+00 C1 OS9 1.05478066 0.787 0.338 0.00E+00 C1 R3HDM2 1.031695733 0.742 0.167 0.00E+00 C1 B3GNT7 1.031632795 0.549 0.088 0.00E+00 C1 MORF4L1 1.023176967 0.768 0.409 0.00E+00 C1 TMEM165 0.999912261 0.649 0.196 0.00E+00 C1 GALNT3 0.995165397 0.705 0.254 0.00E+00 C1 TFF1 0.962929536 0.96 0.591 0.00E+00 C1 LY6D 0.94328396 0.484 0.007 0.00E+00 C1 MIA 0.895355862 0.623 0.103 0.00E+00 C1 MDM2 0.864864363 0.615 0.089 0.00E+00 C1 YEATS4 0.864197638 0.585 0.128 0.00E+00 C1 CPM 0.831257805 -

MOL ID MOL Name OB DL Herb Source 1 MOL011616 Fortunellin
Table S1. Detailed information of active compounds (OB ≥ 30% and DL ≥ 0.18) in PHF MOL ID MOL Name OB DL Herb source 1 MOL011616 Fortunellin 35.65 0.74 BH 2 MOL001689 acacetin 34.97 0.24 BH 3 MOL001790 Linarin 39.84 0.71 BH 4 MOL002881 Diosmetin 31.14 0.27 BH 5 MOL000359 sitosterol 36.91 0.75 BH、DP 6 MOL004328 naringenin 59.29 0.21 BH 7 MOL000471 aloe-emodin 83.38 0.24 BH 8 MOL005190 eriodictyol 71.79 0.24 BH 9 MOL005573 Genkwanin 37.13 0.24 BH 10 MOL000006 luteolin 36.16 0.25 BH、JYH 11 MOL000173 wogonin 30.68 0.23 CZ 2-Hydroxyisoxypropyl-3-hydroxy-7- 12 MOL000179 isopentene-2,3-dihydrobenzofuran-5- 45.2 0.2 CZ carboxylic 13 MOL000184 NSC63551 39.25 0.76 CZ Stigmasterol 3-O-beta-D- 14 MOL000186 43.83 0.76 CZ glucopyranoside_qt 15 MOL000188 3β-acetoxyatractylone 40.57 0.22 CZ 16 MOL000085 beta-daucosterol_qt 36.91 0.75 CZ 17 MOL000088 beta-sitosterol 3-O-glucoside_qt 36.91 0.75 CZ 18 MOL000092 daucosterin_qt 36.91 0.76 CZ 19 MOL000094 daucosterol_qt 36.91 0.76 CZ 20 MOL001925 paeoniflorin_qt 68.18 0.4 DP 21 MOL000211 Mairin 55.38 0.78 DP 22 MOL000422 kaempferol 41.88 0.24 DP、JYH 23 MOL000492 (+)-catechin 54.83 0.24 DP 24 MOL007003 benzoyl paeoniflorin 31.14 0.54 DP 25 MOL007369 4-O-methylpaeoniflorin_qt 67.24 0.43 DP 5-[[5-(4-methoxyphenyl)-2- 26 MOL007374 43.44 0.3 DP furyl]methylene]barbituric acid 27 MOL007382 mudanpioside-h_qt 2 42.36 0.37 DP 28 MOL007384 paeonidanin_qt 65.31 0.35 DP 29 MOL000098 quercetin 46.43 0.28 DP、JYH、HB 30 MOL001454 berberine 36.86 0.78 HB 31 MOL001458 coptisine 30.67 0.86 HB 32 MOL002636 Kihadalactone A 34.21 -

(HBV) Infection of the Liver Ahmed Mohamed Abdel-B Diab Purdue University
Purdue University Purdue e-Pubs Open Access Dissertations Theses and Dissertations January 2016 The oler of PLK1 in Hepatitis B Virus (HBV) infection of the liver Ahmed Mohamed Abdel-B Diab Purdue University Follow this and additional works at: https://docs.lib.purdue.edu/open_access_dissertations Recommended Citation Diab, Ahmed Mohamed Abdel-B, "The or le of PLK1 in Hepatitis B Virus (HBV) infection of the liver" (2016). Open Access Dissertations. 1214. https://docs.lib.purdue.edu/open_access_dissertations/1214 This document has been made available through Purdue e-Pubs, a service of the Purdue University Libraries. Please contact [email protected] for additional information. Graduate School Form 30 Updated 12/26/2015 PURDUE UNIVERSITY GRADUATE SCHOOL Thesis/Dissertation Acceptance This is to certify that the thesis/dissertation prepared By Ahmed Mohamed Abdel-B Diab Entitled The role of PLK1 in Hepatitis B Virus (HBV) Infection of the Liver For the degree of Doctor of Philosophy Is approved by the final examining committee: Ourania Andrisani Andy W. Tao Co-chair Fabien Zoulim Co-chair Robert Geahlen Xiaoqi Liu To the best of my knowledge and as understood by the student in the Thesis/Dissertation Agreement, Publication Delay, and Certification Disclaimer (Graduate School Form 32), this thesis/dissertation adheres to the provisions of Purdue University’s “Policy of Integrity in Research” and the use of copyright material. Approved by Major Professor(s): Ourania Andrisani Laurie Jaeger 10/6/2016 Approved by: Head of the Departmental Graduate Program Date i THE ROLE OF PLK1 IN HEPATITIS B VIRUS (HBV) INFECTION OF THE LIVER A Dissertation Submitted to the Faculty of Purdue University by Ahmed M Diab In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy December 2016 Purdue University West Lafayette, Indiana ii ACKNOWLEDGEMENTS I would like to begin by extending my gratitude to my committee members Robert Geahlen, Andy W. -

(B6;129.Cg-Gt(ROSA)26Sor Tm20(CAG-Ctgf-GFP)Jsd) Were Crossed with Female Foxd1cre/+ Heterozygote Mice 1, and Experimental Mice Were Selected As Foxd1cre/+; Rs26cig/+
Supplemental Information SI Methods Animal studies Heterozygote mice (B6;129.Cg-Gt(ROSA)26Sor tm20(CAG-Ctgf-GFP)Jsd) were crossed with female Foxd1Cre/+ heterozygote mice 1, and experimental mice were selected as Foxd1Cre/+; Rs26CIG/+. In some studies Coll-GFPTg or TCF/Lef:H2B-GFPTg mice or Foxd1Cre/+; Rs26tdTomatoR/+ mice were used as described 2; 3. Left kidneys were subjected to ureteral obstruction using a posterior surgical approach as described 2. In some experiments recombinant mouse DKK1 (0.5mg/kg) or an equal volume of vehicle was administered by daily IP injection. In the in vivo ASO experiment, either specific Lrp6 (TACCTCAATGCGATTT) or scrambled negative control ASO (AACACGTCTATACGC) (30mg/kg) (Exiqon, LNA gapmers) was administered by IP injection on d-1, d1, d4, and d7. In other experiments anti-CTGF domain-IV antibodies (5mg/kg) or control IgG were administered d-1, d1 and d6. All animal experiments were performed under approved IACUC protocols held at the University of Washington and Biogen. Recombinant protein and antibody generation and characterization Human CTGF domain I (sequence Met1 CPDEPAPRCPAGVSLVLDGCGCCRVCAKQLGELCTERDPCDPHKGLFC), domain I+II (sequence Met1CPDEPAPRCPAGVSLVLDGCGCCRVCAKQLGELCTERDPCDPHKGLFCCIFGGT VYRSGESFQSSCKYQCTCLDGAVGCMPLCSMDVRLPSPDCPFPRRVKLPGKCCEE) were cloned and expressed in 293 cells, and purified by Chelating SFF(Ni) Column, tested for single band by SEC and PAGE, and tested for absence of contamination. Domain-IV (sequence GKKCIRTPKISKPIKFELSGCTSMKTYRAKFCGVCTDGRCCTPHRTTTLPVEFKCPDGE VMKKNMMFIKTCACHYNCPGDNDIFESLYYRKMY) was purchased from Peprotech. Mouse or human DKK1 was generated from the coding sequence with some modifications and a tag. Secreted protein was harvested from 293 cells, and purified by nickel column, and tested for activity in a supertopflash (STF) assay 4. DKK1 showed EC50 of 0.69nM for WNT3a-induced WNT signaling in STF cells.