Complete Mitochondrial Genome of the Laced Fritillary Argyreus Hyperbius
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Complete Mitochondrial Genome of Hipparchia Autonoe (Esper, 1783) (Lepidoptera: Nymphalidae): Investigation of Intraspecific Variations on Mitochondrial Genome
Mitochondrial DNA Part B Resources ISSN: (Print) 2380-2359 (Online) Journal homepage: https://www.tandfonline.com/loi/tmdn20 The complete mitochondrial genome of Hipparchia autonoe (Esper, 1783) (Lepidoptera: Nymphalidae): investigation of intraspecific variations on mitochondrial genome Yeong-Don Lee, Jungmo Lee, Do-Sung Kim, Jonghyun Park, Hong Xi, Jeehee Roh, Dong-Soon Kim, Sang June Nam, Seong-Ki Kim, Jin-Young Song & Jongsun Park To cite this article: Yeong-Don Lee, Jungmo Lee, Do-Sung Kim, Jonghyun Park, Hong Xi, Jeehee Roh, Dong-Soon Kim, Sang June Nam, Seong-Ki Kim, Jin-Young Song & Jongsun Park (2020) The complete mitochondrial genome of Hipparchiaautonoe (Esper, 1783) (Lepidoptera: Nymphalidae): investigation of intraspecific variations on mitochondrial genome, Mitochondrial DNA Part B, 5:2, 1542-1544, DOI: 10.1080/23802359.2020.1742230 To link to this article: https://doi.org/10.1080/23802359.2020.1742230 © 2020 The Author(s). Published by Informa Published online: 24 Mar 2020. UK Limited, trading as Taylor & Francis Group. Submit your article to this journal Article views: 95 View related articles View Crossmark data Full Terms & Conditions of access and use can be found at https://www.tandfonline.com/action/journalInformation?journalCode=tmdn20 MITOCHONDRIAL DNA PART B 2020, VOL. 5, NO. 2, 1542–1544 https://doi.org/10.1080/23802359.2020.1742230 MITOGENOME ANNOUNCEMENT The complete mitochondrial genome of Hipparchia autonoe (Esper, 1783) (Lepidoptera: Nymphalidae): investigation of intraspecific variations on mitochondrial -

Evolution of Insect Color Vision: from Spectral Sensitivity to Visual Ecology
EN66CH23_vanderKooi ARjats.cls September 16, 2020 15:11 Annual Review of Entomology Evolution of Insect Color Vision: From Spectral Sensitivity to Visual Ecology Casper J. van der Kooi,1 Doekele G. Stavenga,1 Kentaro Arikawa,2 Gregor Belušic,ˇ 3 and Almut Kelber4 1Faculty of Science and Engineering, University of Groningen, 9700 Groningen, The Netherlands; email: [email protected] 2Department of Evolutionary Studies of Biosystems, SOKENDAI Graduate University for Advanced Studies, Kanagawa 240-0193, Japan 3Department of Biology, Biotechnical Faculty, University of Ljubljana, 1000 Ljubljana, Slovenia; email: [email protected] 4Lund Vision Group, Department of Biology, University of Lund, 22362 Lund, Sweden; email: [email protected] Annu. Rev. Entomol. 2021. 66:23.1–23.28 Keywords The Annual Review of Entomology is online at photoreceptor, compound eye, pigment, visual pigment, behavior, opsin, ento.annualreviews.org anatomy https://doi.org/10.1146/annurev-ento-061720- 071644 Abstract Annu. Rev. Entomol. 2021.66. Downloaded from www.annualreviews.org Copyright © 2021 by Annual Reviews. Color vision is widespread among insects but varies among species, depend- All rights reserved ing on the spectral sensitivities and interplay of the participating photore- Access provided by University of New South Wales on 09/26/20. For personal use only. ceptors. The spectral sensitivity of a photoreceptor is principally determined by the absorption spectrum of the expressed visual pigment, but it can be modified by various optical and electrophysiological factors. For example, screening and filtering pigments, rhabdom waveguide properties, retinal structure, and neural processing all influence the perceived color signal. -

Region of Nymphalidae and Libytheidae (Lepidoptera) from Japan
MORPHOLOGICAL STUDIES ON THE OCCIPITAL REGION OF NYMPHALIDAE AND LIBYTHEIDAE (LEPIDOPTERA) FROM JAPAN Takashi TSUBUKI and Nagao KOYAMA * .h2monji junior High School, 10-33, Kita6tsz{ha 1, Toshima-feu, Toleyo (170), 1opan ** Biological Laboratory, Shinshi2 Udeiversity, Cllada (386), 14pan CONTENTS I. Introduction・--・ny・・・・・・・・・・・・・・・・・-・-・・・・・・-・---・--t・-・---・ny・・・・-・-・・・-・-・・-・・・1 II. Materials and methods・-・・・・・・・・・t・・・・・・t・・・・・・・・・・・・・・`・・・・・・・・・・-・・・-・・・・・・-d・・・・・・・・・・・・2 III. General morphology of the occipital region ・・・・・・}・・・・・・・・・・・・・・・・・・・・・・・・・・・・・4 IV. Occipital structure of each species in Nymphaliclae and Libytheidae ・-ib-・・・・-・・・-・ny・・・・・--・-・・・・・・・-・・-・----・b-・・・----・-・・・-・・-・--・・-6 V. Phylogenical grouping of Nymphalidae based on the occipital structure・t・・・・・・・--・--・・-・-・・・・・・・・・・-・・・・-ny-・・・ny-・--・-----・H"""""".","".."."lg VI. Summary ・・・・-・-・-・・・・・・-・・----・・・-・・・・-・-・-・-----・・-------・-・----・23 VII. Literatures cited ・・・--・・・・・・・・・`・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・--・-・・-・・・・・・tt・・・・・・23 I. INTRODUCTION A considerable number of studies have been carried out on the occipital morphology of moths since YAGI and KoYAMA's report of 1963 (KoYAMA and MlyATA, 1969, 1970, 1975;MIYATA, 1971, 1972, 1973, 1974, 1975;MIyATA and KOYAMA, 1971, 1972, 1976). In the butterfiies, however, few papers were available concerning the occipital region, on which EHRucH (1958, a b), YAGI and KoyAMA (1963) and KoyAMA and OGAwA (1972) briefly described. Then, tried to study the occipital region of butterflies, the authors observed it preliminarily (TsuBuKI et aL 1975). The present paper deals with the occipital structure of Nymphalidae and Libytheidae from Japan with reference to its bearing on systematics. Before going further, the authors wish to express their gratitude to Prof. Dr. H. SAwADA and Prof. Dr. N. GOKAN, Tokyo University of Agriculture, for their advice and assistance. Thanks are also due to Mr. N. K6DA who gave valuable materials for this work, and to Mr. Y. YAGucm and the members of JEOL Ltd. -
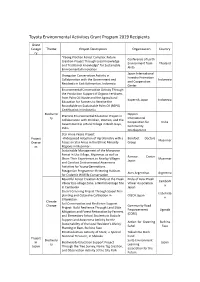
List of Previous Grant Projects
Toyota Environmental Activities Grant Program 2019 Recipients Grant Catego Theme Project Description Organization Country ry "Kaeng Krachan Forest Complex: Future Conference of Earth Creation Project Through Local Knowledge Environment from Thailand and Traditional Knowledge" for Sustainable Akita Environmental Innovation Japan International Orangutan Conservation Activity in Forestry Promotion Collaboration with the Government and Indonesia and Cooperation Residents in East Kalimantan, Indonesia Center Environmental Conservation Activity Through the Production Support of Organic Fertilizers from Palm Oil Waste and the Agricultural Kopernik Japan Indonesia Education for Farmers to Receive the Roundtable on Sustainable Palm Oil (RSPO) Certification in Indonesia Biodiversi Nippon Practical Environmental Education Project in ty International Collaboration with Children, Women, and the Cooperation for India Government in a Rural Village in Bodh Gaya, Community India Development Star Anise Peace Project Project -Widespread Adoption of Agroforestry with a Barefoot Doctors Myanmar Overse Focus on Star Anise in the Ethnic Minority Group as Regions in Myanmar- Sustainable Management of the Mangrove Forest in Uto Village, Myanmar, as well as Ramsar Center Share Their Experiences to Nearby Villages Myanmar Japan and Conduct Environmental Awareness Activities for Young Generations Patagonian Programme: Restoring Habitats Aves Argentinas Argentina for Endemic Wildlife Conservation Beautiful Forest Creation Activity at the Preah Pride of Asia: Preah -

Practical Experiences in Invasive Alien Plant Control
ROSALIA Handbooks ROSALIA Handbooks Practical Experiences in Invasive Alien Plant Control Second, revised and expanded edition Invasive plant species pose major agricultural, silvicultural, human health and ecological problems worldwide, and are considered the most signifi cant threat for nature conservation. Species invading natural areas in Hungary have been described by a number of books published in the Practical Experiences in Invasive Alien Plant Control last few years. A great amount of experience has been gathered about the control of these species in some areas, which we can read about in an increasing number of articles; however, no book has been published with regards to the whole country. Invasions affecting larger areas require high energy and cost input, and the effectiveness and successfulness of control can be infl uenced by a number of factors. The development of effective, widely applicable control and eradication technologies is preceded by experiments and examinations which are based on a lot of practical experience and often loaded with negative experiences. National park directorates, forest and agricultural managers and NGOs in many parts of Hungary are combatting the spread of invasive species; however, the exchange of information and conclusion of experiences among the managing bodies is indispensable. The aim of the present volume is to facilitate this by summarizing experiences and the methods applied in practice; which, we hope, will enable us to successfully stop the further spread of invasive plant species and effectively protect our natural values. Magyarország-Szlovákia Partnerséget építünk Határon Átnyúló Együttműködési Program 2007-2013 Duna-Ipoly National Park Directorate rrosaliaosalia kkezikonyvezikonyv 3 aangng jjav.inddav.indd 1 22017.12.15.017.12.15. -

The Taxonomy of the Japanese Oak Red Scale Insect
Zootaxa 3630 (2): 291–307 ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ Article ZOOTAXA Copyright © 2013 Magnolia Press ISSN 1175-5334 (online edition) http://dx.doi.org/10.11646/zootaxa.3630.2.5 http://zoobank.org/urn:lsid:zoobank.org:pub:373402BA-36A9-4323-AE9D-A84D89C47231 The taxonomy of the Japanese oak red scale insect, Kuwania quercus (Kuwana) (Hemiptera: Coccoidea: Kuwaniidae), with a generic diagnosis, a key to species and description of a new species from California SAN’AN WU1, NAN NAN1, PENNY GULLAN2 & JUN DENG1 1The Key Laboratory for Silviculture and Conservation of Ministry of Education, Beijing Forestry University, Beijing, P. R. China 100083. E-mail: [email protected] 2Division of Evolution, Ecology and Genetics, Research School of Biology, The Australian National University, Canberra, A.C.T. 0200, Australia. E-mail: [email protected] Abstract The oak red scale insect, Kuwania quercus (Kuwana), was described from specimens collected from the bark of oak trees (Quercus species) in Japan. More recently, the species has been identified from California and China, but Californian specimens differ morphologically from Japanese material and are considered here to be a new species based on both morphological and molecular data. In this paper, an illustrated redescription of K. quercus is provided based on type specimens consisting of adult females, first-instar nymphs and intermediate-stage females, and a lectotype is designated for Sasakia quercus Kuwana. The new Californian species, Kuwania raygilli Wu & Gullan, is described and illustrated based on the adult female, first-instar nymph and intermediate-stage female. A new generic diagnosis for Kuwania Cockerell based on adult females and first-instar nymphs, and a key to species based on adult females are included. -

Photonic Crystal Structure of Wing Scales in Sasakia Charonda Butterflies
Materials Transactions, Vol. 51, No. 2 (2010) pp. 202 to 208 Special Issue on Development and Fabrication of Advanced Materials Assisted by Nanotechnology and Microanalysis #2010 The Japan Institute of Metals Photonic Crystal Structure of Wing Scales in Sasakia Charonda Butterflies Jirˇina Mateˇjkova´-Plsˇkova´1, Dalibor Jancˇik1, Miroslav Masˇla´nˇ1, Satoshi Shiojiri2 and Makoto Shiojiri3;* 1Centre for Nanomaterial Research, Faculty of Science, Palacky University in Olomouc, Slechtitelu 11, 783 71 Olomouc, Czech Republic 2Matsubara Junior High-school, Kyoto 604-8812, Japan 3Professor Emeritus of Kyoto Institute of Technology, 1-297 Wakiyama, Kyoto 618-0091, Japan The hindwings of the male Sasakia charonda charonda butterflies comprise iridescent purple-blue areas, iridescent white-pearl areas, yellow spots and red spots as well as brown background. We have examined the microstructure of their scales by scanning electron microscopy, for applying their photonic crystal structures to fine light manipulators such as reflection elements in laser diodes. The scales in the yellow spots, red spots and brown background have almost the same structure, which is an optical diffraction grating made of ridges with two cuticle layers. Their difference comes from the contained pigments. The scales in the iridescent purple-blue and white-pearl are also the same in structure. They have seven tilted cuticle layers lapped on the ridges, which also constitute a grating. The widths of the ridge and groove in the grating are different between scales of the two kinds. It is shown that the vivid iridescence is mainly attributed to multiple interferences caused between rays reflected from the seven cuticle layers with air gaps. -

Analysis of Biodiversity Offset for Road Projects in Japan
Analysis of Biodiversity Offset for Road Projects in Japan Hideyuki ITO Jun NISHIJIMA Department of Transportation Systems Department of Transportation Systems Engineering, College of Science and Engineering, College of Science and Technology, Nihon University, Japan Technology, Nihon University, Japan Takahiro FUJII Makoto Ooba Department of Transportation Systems Center for Social and Environmental Systems Engineering, College of Science and Research, Technology, Nihon University, Japan National Institute for Environmental Studies, Japan Kiichiro HAYASHI EcoTopia Science Institute, Nagoya University, Japan Abstract: In Japan, Environmental Impact Assessment Law was enforced in 1999. This law required developers to consider how to avoid, reduce, and compensate the adverse impacts on natural environment under the purview of mitigation hierarchy. However, whether biodiversity offset projects are implemented or not depends on developers. In addition, various biodiversity offset projects have been conducted by developers through trial and error because the assessment and planning method of biodiversity offset has not been established in Japan. Therefore, the purpose of this research is to analyze the current situation regarding biodiversity offset projects focusing on road projects that was applied Environmental Impact Assessment Law all over Japan from various viewpoints such as maintenance, monitoring, cost, and public involvement as well as the planning methods of biodiversity offset projects. The analysis was performed through a questionnaire administered to developers and field surveys were conducted at some offset sites. The reason why authors focused on road projects is because there are many biodiversity offset cases among development projects. Consequently, this research could clarify the recent implementation method of biodiversity offset projects for road projects and highlights the issues and future tasks. -

TPG Index Volumes 1-35 1986-2020
Public Garden Index – Volumes 1-35 (1986 – 2020) #Giving Tuesday. HOW DOES YOUR GARDEN About This Issue (continued) GROW ? Swift 31 (3): 25 Dobbs, Madeline (continued) #givingTuesday fundraising 31 (3): 25 Public garden management: Read all #landscapechat about it! 26 (W): 5–6 Corona Tools 27 (W): 8 Rocket science leadership. Interview green industry 27 (W): 8 with Elachi 23 (1): 24–26 social media 27 (W): 8 Unmask your garden heroes: Taking a ValleyCrest Landscape Companies 27 (W): 8 closer look at earned revenue. #landscapechat: Fostering green industry 25 (2): 5–6 communication, one tweet at a time. Donnelly, Gerard T. Trees: Backbone of Kaufman 27 (W): 8 the garden 6 (1): 6 Dosmann, Michael S. Sustaining plant collections: Are we? 23 (3/4): 7–9 AABGA (American Association of Downie, Alex. Information management Botanical Gardens and Arboreta) See 8 (4): 6 American Public Gardens Association Eberbach, Catherine. Educators without AABGA: The first fifty years. Interview by borders 22 (1): 5–6 Sullivan. Ching, Creech, Lighty, Mathias, Eirhart, Linda. Plant collections in historic McClintock, Mulligan, Oppe, Taylor, landscapes 28 (4): 4–5 Voight, Widmoyer, and Wyman 5 (4): 8–12 Elias, Thomas S. Botany and botanical AABGA annual conference in Essential gardens 6 (3): 6 resources for garden directors. Olin Folsom, James P. Communication 19 (1): 7 17 (1): 12 Rediscovering the Ranch 23 (2): 7–9 AAM See American Association of Museums Water management 5 (3): 6 AAM accreditation is for gardens! SPECIAL Galbraith, David A. Another look at REPORT. Taylor, Hart, Williams, and Lowe invasives 17 (4): 7 15 (3): 3–11 Greenstein, Susan T. -

The Taxonomy of the Japanese Oak Red Scale Insect
Zootaxa 3630 (2): 291–307 ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ Article ZOOTAXA Copyright © 2013 Magnolia Press ISSN 1175-5334 (online edition) http://dx.doi.org/10.11646/zootaxa.3630.2.5 http://zoobank.org/urn:lsid:zoobank.org:pub:373402BA-36A9-4323-AE9D-A84D89C47231 The taxonomy of the Japanese oak red scale insect, Kuwania quercus (Kuwana) (Hemiptera: Coccoidea: Kuwaniidae), with a generic diagnosis, a key to species and description of a new species from California SAN’AN WU1, NAN NAN1, PENNY GULLAN2 & JUN DENG1 1The Key Laboratory for Silviculture and Conservation of Ministry of Education, Beijing Forestry University, Beijing, P. R. China 100083. E-mail: [email protected] 2Division of Evolution, Ecology and Genetics, Research School of Biology, The Australian National University, Canberra, A.C.T. 0200, Australia. E-mail: [email protected] Abstract The oak red scale insect, Kuwania quercus (Kuwana), was described from specimens collected from the bark of oak trees (Quercus species) in Japan. More recently, the species has been identified from California and China, but Californian specimens differ morphologically from Japanese material and are considered here to be a new species based on both morphological and molecular data. In this paper, an illustrated redescription of K. quercus is provided based on type specimens consisting of adult females, first-instar nymphs and intermediate-stage females, and a lectotype is designated for Sasakia quercus Kuwana. The new Californian species, Kuwania raygilli Wu & Gullan, is described and illustrated based on the adult female, first-instar nymph and intermediate-stage female. A new generic diagnosis for Kuwania Cockerell based on adult females and first-instar nymphs, and a key to species based on adult females are included. -

Practical Experiences in Invasive Alien Plant Control. Rosalia Handbooks
ROSALIA Handbooks ROSALIA Handbooks Practical Experiences in Invasive Alien Plant Control Invasive plant species pose major agricultural, silvicultural, human health and ecological problems worldwide, and are considered the most signifi cant threat for nature conservation. Species invading natural areas in Hungary have been described by a number of books published in the Practical Experiences in Invasive Alien Plant Control last few years. A great amount of experience has been gathered about the control of these species in some areas, which we can read about in an increasing number of articles; however, no book has been published with regards to the whole country. Invasions affecting larger areas require high energy and cost input, and the effectiveness and successfulness of control can be infl uenced by a number of factors. The development of effective, widely applicable control and eradication technologies is preceded by experiments and examinations which are based on a lot of practical experience and often loaded with negative experiences. National park directorates, forest and agricultural managers and NGOs in many parts of Hungary are combatting the spread of invasive species; however, the exchange of information and conclusion of experiences among the managing bodies is indispensable. The aim of the present volume is to facilitate this by summarizing experiences and the methods applied in practice; which, we hope, will enable us to successfully stop the further spread of invasive plant species and effectively protect our natural values. Hungary-Slovakia Cross-border Co-operation Programme 2007-2013 Duna-Ipoly National Park Directorate Financial support for this manual has been provided by “Unified protection against invasive alien plants in sand and floodplain habitats” project. -

A RESEARCH on Libythea Celtis (LAICHARTING, 1782) (LEPIDOPTERA, NYMPHALIDAE) NETTLE-TREE BUTTERFLY in DEVREK, TURKEY
ZKÜ Bartın Orman Fakültesi Dergisi Yıl: 2006 Cilt:8 Sayı:9 A RESEARCH ON Libythea celtis (LAICHARTING, 1782) (LEPIDOPTERA, NYMPHALIDAE) NETTLE-TREE BUTTERFLY IN DEVREK, TURKEY Azize TOPER KAYGIN, Hilmi SÖNMEZYILDIZ, Yafes YILDIZ ZKÜ Bartın Orman Fakültesi, Orman Mühendisliği Bölümü ABSTRACT This research has been carried out in 2005, in Devrek. For collection of plants and larvae from the field and for breeding larva in laboratory, identification of insects and preserving technique modern methods were used. In our investigations in spring 2005, interestingly we found Libythea celtis (Laicharting, 1782)’s larvae were feeding on foliage of Alnus glutinosa subsp. glutinosa although Celtis sp. and essentially Celtis australis are foodplant of this butterfly. Keywords : Libythea celtis, Devrek, Alnus, Celtis. DEVREK’TE ÇİTLENBİK KELEBEĞİ LIBYTHEA CELTIS (LAICHARTING, 1782) (LEPIDOPTERA, NYMPHALIDAE) ÜZERİNE BİR ARAŞTIRMA ÖZET Bu araştırma 2005 yılında Devrek’te gerçekleştirilmiştir. Bitki örneklerinin ve larvaların toplanması, larvaların laboratuarda beslenmesi, erginlerin elde edilmesi, teşhisi ve koleksiyonda muhafaza altına alınmasında modern yöntemler uygulanmıştır. 2005 yılı bahar aylarında yapılan araştırmada, literatürde Libythea celtis (Laicharting, 1782)’in konukçusu Celtis türleri ve özellikle Celtis australis olarak belirtilmesine rağmen, bu kelebeğin larvalarının Alnus glutinosa subsp. glutinosa’nın yaprakları üzerinde beslendiği tespit edilmiştir. Anahtar kelimeler: Libythea celtis, Devrek, Alnus, Celtis. 1. INTRODUCTION L. celtis is strange and rare butterfly (wingspans is about 34-44 mm). It has unusual wings and a long palpi. So, it is distinguishable by its long snout and its projection on the front wing. It is the only one its subfamily (Nymphalidae, Libytheinae) in Western Europe. Its name is tengu-cho in Japanese http://pro.tok2.com/~tokyonature).