Role of Specialized Composition of SWI/SNF Complexes in Prostate Cancer Lineage Plasticity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7, -

The Role of Inhibitors of Differentiation Proteins ID1 and ID3 in Breast Cancer Metastasis
The role of Inhibitors of Differentiation proteins ID1 and ID3 in breast cancer metastasis Wee Siang Teo A thesis in fulfilment of the requirements for the degree of Doctor of Philosophy St Vincent’s Clinical School, Faculty of Medicine The University of New South Wales Cancer Research Program The Garvan Institute of Medical Research Sydney, Australia March, 2014 THE UNIVERSITY OF NEW SOUTH WALES Thesis/Dissertation Sheet Surname or Family name: Teo First name: Wee Siang Abbreviation for degree as given in the University calendar: PhD (Medicine) School: St Vincent’s Clinical School Faculty: Faculty of Medicine Title: The role of Inhibitors of Differentiation proteins ID1 and ID3 in breast cancer metastasis Abstract 350 words maximum: (PLEASE TYPE) Breast cancer is a leading cause of cancer death in women. While locally-confined breast cancer is generally curable, the survival of patients with metastatic breast cancer is very poor. Treatment for metastatic breast cancer is palliative not curative due to the lack of targeted therapies. Metastasis is a complex process that still remains poorly understood, thus a detailed understanding of the biological complexity that underlies breast cancer metastasis is essential in reducing the lethality of this disease. The Inhibitor of Differentiation proteins 1 and 3 (ID1/3) are transcriptional regulators that control many cell fate and developmental processes and are often deregulated in cancer. ID1/3 are required and sufficient for the metastasis of breast cancer in experimental models. However, the mechanisms by which ID1/3 mediate metastasis in breast cancer remain to be determined. Little is known about pathways regulated by ID1/3 in breast cancer as well as their functional role in the multiple steps of metastatic progression. -

Loss of the Neural-Specific BAF Subunit ACTL6B Relieves Repression of Early Response Genes and Causes Recessive Autism
Loss of the neural-specific BAF subunit ACTL6B relieves repression of early response genes and causes recessive autism Wendy Wenderskia,b,c,d, Lu Wange,f,g,1, Andrey Krokhotina,b,c,d,1, Jessica J. Walshh, Hongjie Lid,i, Hirotaka Shojij, Shereen Ghoshe,f,g, Renee D. Georgee,f,g, Erik L. Millera,b,c,d, Laura Eliasa,b,c,d, Mark A. Gillespiek, Esther Y. Sona,b,c,d, Brett T. Staahla,b,c,d, Seung Tae Baeke,f,g, Valentina Stanleye,f,g, Cynthia Moncadaa,b,c,d, Zohar Shiponya,b,c,d, Sara B. Linkerl, Maria C. N. Marchettol, Fred H. Gagel, Dillon Chene,f,g, Tipu Sultanm, Maha S. Zakin, Jeffrey A. Ranishk, Tsuyoshi Miyakawaj, Liqun Luod,i, Robert C. Malenkah, Gerald R. Crabtreea,b,c,d,2, and Joseph G. Gleesone,f,g,2 aDepartment of Pathology, Stanford Medical School, Palo Alto, CA 94305; bDepartment of Genetics, Stanford Medical School, Palo Alto, CA 94305; cDepartment of Developmental Biology, Stanford Medical School, Palo Alto, CA 94305; dHoward Hughes Medical Institute, Stanford University, Palo Alto, CA 94305; eDepartment of Neuroscience, University of California San Diego, La Jolla, CA 92037; fHoward Hughes Medical Institute, University of California San Diego, La Jolla, CA 92037; gRady Children’s Institute of Genomic Medicine, University of California San Diego, La Jolla, CA 92037; hNancy Pritztker Laboratory, Department of Psychiatry and Behavioral Sciences, Stanford Medical School, Palo Alto, CA 94305; iDepartment of Biology, Stanford University, Palo Alto, CA 94305; jDivision of Systems Medical Science, Institute for Comprehensive Medical Science, Fujita Health University, 470-1192 Toyoake, Aichi, Japan; kInstitute for Systems Biology, Seattle, WA 98109; lLaboratory of Genetics, The Salk Institute for Biological Studies, La Jolla, CA 92037; mDepartment of Pediatric Neurology, Institute of Child Health, Children Hospital Lahore, 54000 Lahore, Pakistan; and nClinical Genetics Department, Human Genetics and Genome Research Division, National Research Centre, 12311 Cairo, Egypt Edited by Arthur L. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

The Proteasomal Deubiquitinating Enzyme PSMD14 Regulates Macroautophagy by Controlling Golgi-To-ER Retrograde Transport
Supplementary Materials The proteasomal deubiquitinating enzyme PSMD14 regulates macroautophagy by controlling Golgi-to-ER retrograde transport Bustamante HA., et al. Figure S1. siRNA sequences directed against human PSMD14 used for Validation Stage. Figure S2. Primer pairs sequences used for RT-qPCR. Figure S3. The PSMD14 DUB inhibitor CZM increases the Golgi apparatus area. Immunofluorescence microscopy analysis of the Golgi area in parental H4 cells treated for 4 h either with the vehicle (DMSO; Control) or CZM. The Golgi marker GM130 was used to determine the region of interest in each condition. Statistical significance was determined by Student's t-test. Bars represent the mean ± SEM (n =43 cells). ***P <0.001. Figure S4. CZM causes the accumulation of KDELR1-GFP at the Golgi apparatus. HeLa cells expressing KDELR1-GFP were either left untreated or treated with CZM for 30, 60 or 90 min. Cells were fixed and representative confocal images were acquired. Figure S5. Effect of CZM on proteasome activity. Parental H4 cells were treated either with the vehicle (DMSO; Control), CZM or MG132, for 90 min. Protein extracts were used to measure in vitro the Chymotrypsin-like peptidase activity of the proteasome. The enzymatic activity was quantified according to the cleavage of the fluorogenic substrate Suc-LLVY-AMC to AMC, and normalized to that of control cells. The statistical significance was determined by One-Way ANOVA, followed by Tukey’s test. Bars represent the mean ± SD of biological replicates (n=3). **P <0.01; n.s., not significant. Figure S6. Effect of CZM and MG132 on basal macroautophagy. (A) Immunofluorescence microscopy analysis of the subcellular localization of LC3 in parental H4 cells treated with either with the vehicle (DMSO; Control), CZM for 4 h or MG132 for 6 h. -
Drosophila and Human Transcriptomic Data Mining Provides Evidence for Therapeutic
Drosophila and human transcriptomic data mining provides evidence for therapeutic mechanism of pentylenetetrazole in Down syndrome Author Abhay Sharma Institute of Genomics and Integrative Biology Council of Scientific and Industrial Research Delhi University Campus, Mall Road Delhi 110007, India Tel: +91-11-27666156, Fax: +91-11-27662407 Email: [email protected] Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 Running head: Pentylenetetrazole mechanism in Down syndrome 1 Abstract Pentylenetetrazole (PTZ) has recently been found to ameliorate cognitive impairment in rodent models of Down syndrome (DS). The mechanism underlying PTZ’s therapeutic effect is however not clear. Microarray profiling has previously reported differential expression of genes in DS. No mammalian transcriptomic data on PTZ treatment however exists. Nevertheless, a Drosophila model inspired by rodent models of PTZ induced kindling plasticity has recently been described. Microarray profiling has shown PTZ’s downregulatory effect on gene expression in fly heads. In a comparative transcriptomics approach, I have analyzed the available microarray data in order to identify potential mechanism of PTZ action in DS. I find that transcriptomic correlates of chronic PTZ in Drosophila and DS counteract each other. A significant enrichment is observed between PTZ downregulated and DS upregulated genes, and a significant depletion between PTZ downregulated and DS dowwnregulated genes. Further, the common genes in PTZ Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 downregulated and DS upregulated sets show enrichment for MAP kinase pathway. My analysis suggests that downregulation of MAP kinase pathway may mediate therapeutic effect of PTZ in DS. Existing evidence implicating MAP kinase pathway in DS supports this observation. -

(2) Patent Application Publication (10) Pub. No.: US 2011/0023143 A1 Weinstein Et Al
US 2011 00231.43A1 (19) United States (2) Patent Application Publication (10) Pub. No.: US 2011/0023143 A1 Weinstein et al. (43) Pub. Date: Jan. 27, 2011 (54) GENOMIC EDITING OF 2010, provisional application No. 61/308,089, filed on NEURODEVELOPMENTAL GENES IN Feb. 25, 2010, provisional application No. 61/336,000, ANIMALS filed on Jan. 14, 2010, provisional application No. 61/263,904, filed on Nov. 24, 2009, provisional appli (75) Inventors: Edward Weinstein, St. Louis, MO cation No. 61/263,696, filed on Nov. 23, 2009, provi (US); Xiaoxia Cui, St. Louis, MO sional application No. 61/245,877, filed on Sep. 25, (US); Phil Simmons, St. Louis, MO 2009, provisional application No. 61/232,620, filed on (US) Aug. 10, 2009, provisional application No. 61/228, 419, filed on Jul. 24, 2009, provisional application No. Correspondence Address: 61/200,985, filed on Dec. 4, 2008, provisional appli POLSINELLISHUGHART PC cation No. 61/205,970, filed on Jan. 26, 2009. 700 W. 47TH STREET, SUITE 1000 KANSAS CITY, MO 64112-1802 (US) Publication Classification (51) Int. Cl. (73) Assignee: SIGMA-ALDRICH CO., St. G0IN 33/00 (2006.01) Louis, MO (US) A0IR 67/00 (2006.01) (21) Appl. No.: 12/842,578 CI2N 5/10 (2006.01) (52) U.S. Cl. ................... 800/3; 800/13; 800/15; 800/16; (22) Filed: Jul. 23, 2010 800/17: 800/14; 435/325; 435/351; 435/350: 435/366; 435/363; 435/352; 435/353 Related U.S. Application Data (57) ABSTRACT (63) Continuation-in-part of application No. 12/592,852, filed on Dec. -

Abcam Overview of Pcg and Trxg Complexes to See Available Products
Click Gene name or AbID abcam Overview of PcG and TrxG complexes to see available products Gene name Alternative name Known function AbID Species Application Histone modifications H2AK119ub – – ab193203 Hu, Ms, Rt ICC/IF, WB, PepArr H3K4me – – ab176877 Hu, Ms, Rt ChIP, ICC/IF, IHC-P, WB, PepArr H3K4me2 – – ab32356 Hu, Ms, Rt, Chicken, Cow ChIP-seq, ChIP, Flow Cyt, IHC-P, ICC/IF, IP, WB H3K4me3 – – ab213224 Hu, Ms, Rt ChIP-seq, ChIP, Flow Cyt, ICC/IF, IP, WB, Dot blot, PepArr H3K27me3 – – ab192985 Hu, Ms, Rt ChIP-seq, ChIP, IHC-P, ICC/IF, ELISA, WB, PepArr H3K36me3 – – – – – PcG Complex Components Core PRC1 Complex RING1 RING1A ab180170 Hu ICC/IF, IHC-P, WB, IP H2AK119 ubiquitylation RNF2 RING1B ab181140 Hu, Ms, Rt Flow Cyt, ICC/IF, IHC-P, WB PCGF1 – ab259943 Hu, Ms, Rt Flow Cyt, IP, WB PCGF2 MEL18 – – – PCGF3 – – – – H2AK119 ubiquitylation, oligomerization PCGF4 BMI1 ab126783 Hu, Rt ICC/IF, IHC-P, IP, WB PCGF5 – ab201511 Hu, Ms, Rt WB PCGF6 – ab200038 Ms, Rt ICC/IF, IP, WB Canonical PRC1 CBX2 – – – – CBX4 – ab242149 Hu, Ms, Rt ChIP, Flow Cyt, IHC-P, IP, WB CBX6 – H3K27me3 binding ab259848 Hu, Ms, Rt WB, IHC-P CBX7 – ab178411 Hu Flow Cyt, WB CBX8 – ab259849 Hu, Ms, Rt WB PHC1 EDR1 – – – PHC2 – – – – oligomerization/protein-protein interaction PHC3 – ab180577 Hu, Ms, Rt WB SCMH1 – – – – Overview of PcG and TrxG complexes and their core subunits, functions, and recommended recombinant monoclonal antibodies www.abcam.com 1 Gene name Alternative name Known function AbID Species Application Non-canonical PRC1 RYBP YAF2 DNA binding ab185971 -
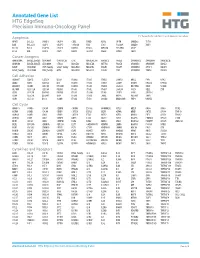
Annotated Gene List HTG Edgeseq Precision Immuno-Oncology Panel
Annotated Gene List HTG EdgeSeq Precision Immuno-Oncology Panel For Research Use Only. Not for use in diagnostic procedures. Apoptosis APAF1 BCL2L1 CARD11 CASP4 CD5L FADD KSR2 OPTN SAMD12 TCF19 BAX BCL2L11 CASP1 CASP5 CORO1A FAS LRG1 PLA2G6 SAMD9 XAF1 BCL10 BCL6 CASP10 CASP8 DAPK2 FASLG MECOM PYCARD SPOP BCL2 BID CASP3 CAV1 DAPL1 GLIPR1 MELK RIPK2 TBK1 Cancer Antigens ANKRD30A BAGE2_BAGE3 CEACAM6 CTAG1A_1B LIPE MAGEA3_A6 MAGEC2 PAGE3 SPANXACD SPANXN4 XAGE1B_1E ARMCX6 BAGE4_BAGE5 CEACAM8 CTAG2 MAGEA1 MAGEA4 MTFR2 PAGE4 SPANXB1 SPANXN5 XAGE2 BAGE CEACAM1 CT45_family GAGE_family MAGEA10 MAGEB2 PAGE1 PAGE5 SPANXN1 SYCP1 XAGE3 BAGE_family CEACAM5 CT47_family HPN MAGEA12 MAGEC1 PAGE2 PBK SPANXN3 TEX14 XAGE5 Cell Adhesion ADAM17 CDH15 CLEC5A DSG3 ICAM2 ITGA5 ITGB2 LAMC3 MBL2 PVR UPK2 ADD2 CDH5 CLEC6A DST ICAM3 ITGA6 ITGB3 LAMP1 MTDH RRAS2 UPK3A ADGRE5 CLDN3 CLEC7A EPCAM ICAM4 ITGAE ITGB4 LGALS1 NECTIN2 SELE VCAM1 ALCAM CLEC12A CLEC9A FBLN1 ITGA1 ITGAL ITGB7 LGALS3 OCLN SELL ZYX CD63 CLEC2B DIAPH3 FXYD5 ITGA2 ITGAM ITLN2 LYVE1 OLR1 SELPLG CD99 CLEC4A DLGAP5 IBSP ITGA3 ITGAX JAML M6PR PECAM1 THY1 CDH1 CLEC4C DSC3 ICAM1 ITGA4 ITGB1 L1CAM MADCAM1 PKP1 UNC5D Cell Cycle ANAPC1 CCND3 CDCA5 CENPH CNNM1 ESCO2 HORMAD2 KIF2C MELK ORC6 SKA3 TPX2 ASPM CCNE1 CDCA8 CENPI CNTLN ESPL1 IKZF1 KIF4A MND1 PATZ1 SP100 TRIP13 AURKA CCNE2 CDK1 CENPL CNTLN ETS1 IKZF2 KIF5C MYBL2 PIF1 SP110 TROAP AURKB CCNF CDK4 CENPU DBF4 ETS2 IKZF3 KIFC1 NCAPG PIMREG SPC24 TUBB BEX1 CDC20 CDK6 CENPW E2F2 EZH2 IKZF4 KNL1 NCAPG2 PKMYT1 SPC25 ZWILCH BEX2 CDC25A CDKN1A CEP250 E2F7 GADD45GIP1 -

Downregulation of Carnitine Acyl-Carnitine Translocase by Mirnas
Page 1 of 288 Diabetes 1 Downregulation of Carnitine acyl-carnitine translocase by miRNAs 132 and 212 amplifies glucose-stimulated insulin secretion Mufaddal S. Soni1, Mary E. Rabaglia1, Sushant Bhatnagar1, Jin Shang2, Olga Ilkayeva3, Randall Mynatt4, Yun-Ping Zhou2, Eric E. Schadt6, Nancy A.Thornberry2, Deborah M. Muoio5, Mark P. Keller1 and Alan D. Attie1 From the 1Department of Biochemistry, University of Wisconsin, Madison, Wisconsin; 2Department of Metabolic Disorders-Diabetes, Merck Research Laboratories, Rahway, New Jersey; 3Sarah W. Stedman Nutrition and Metabolism Center, Duke Institute of Molecular Physiology, 5Departments of Medicine and Pharmacology and Cancer Biology, Durham, North Carolina. 4Pennington Biomedical Research Center, Louisiana State University system, Baton Rouge, Louisiana; 6Institute for Genomics and Multiscale Biology, Mount Sinai School of Medicine, New York, New York. Corresponding author Alan D. Attie, 543A Biochemistry Addition, 433 Babcock Drive, Department of Biochemistry, University of Wisconsin-Madison, Madison, Wisconsin, (608) 262-1372 (Ph), (608) 263-9608 (fax), [email protected]. Running Title: Fatty acyl-carnitines enhance insulin secretion Abstract word count: 163 Main text Word count: 3960 Number of tables: 0 Number of figures: 5 Diabetes Publish Ahead of Print, published online June 26, 2014 Diabetes Page 2 of 288 2 ABSTRACT We previously demonstrated that micro-RNAs 132 and 212 are differentially upregulated in response to obesity in two mouse strains that differ in their susceptibility to obesity-induced diabetes. Here we show the overexpression of micro-RNAs 132 and 212 enhances insulin secretion (IS) in response to glucose and other secretagogues including non-fuel stimuli. We determined that carnitine acyl-carnitine translocase (CACT, Slc25a20) is a direct target of these miRNAs.