Zinforo, INN-Ceftaroline Fosamil
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Medical Review(S) Clinical Review
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 200327 MEDICAL REVIEW(S) CLINICAL REVIEW Application Type NDA Application Number(s) 200327 Priority or Standard Standard Submit Date(s) December 29, 2009 Received Date(s) December 30, 2009 PDUFA Goal Date October 30, 2010 Division / Office Division of Anti-Infective and Ophthalmology Products Office of Antimicrobial Products Reviewer Name(s) Ariel Ramirez Porcalla, MD, MPH Neil Rellosa, MD Review Completion October 29, 2010 Date Established Name Ceftaroline fosamil for injection (Proposed) Trade Name Teflaro Therapeutic Class Cephalosporin; ß-lactams Applicant Cerexa, Inc. Forest Laboratories, Inc. Formulation(s) 400 mg/vial and 600 mg/vial Intravenous Dosing Regimen 600 mg every 12 hours by IV infusion Indication(s) Acute Bacterial Skin and Skin Structure Infection (ABSSSI); Community-acquired Bacterial Pneumonia (CABP) Intended Population(s) Adults ≥ 18 years of age Template Version: March 6, 2009 Reference ID: 2857265 Clinical Review Ariel Ramirez Porcalla, MD, MPH Neil Rellosa, MD NDA 200327: Teflaro (ceftaroline fosamil) Table of Contents 1 RECOMMENDATIONS/RISK BENEFIT ASSESSMENT ......................................... 9 1.1 Recommendation on Regulatory Action ........................................................... 10 1.2 Risk Benefit Assessment.................................................................................. 10 1.3 Recommendations for Postmarketing Risk Evaluation and Mitigation Strategies ........................................................................................................................ -

Use of Ceftaroline Fosamil in Children: Review of Current Knowledge and Its Application
Infect Dis Ther (2017) 6:57–67 DOI 10.1007/s40121-016-0144-8 REVIEW Use of Ceftaroline Fosamil in Children: Review of Current Knowledge and its Application Juwon Yim . Leah M. Molloy . Jason G. Newland Received: November 10, 2016 / Published online: December 30, 2016 Ó The Author(s) 2016. This article is published with open access at Springerlink.com ABSTRACT infections, CABP caused by penicillin- and ceftriaxone-resistant S. pneumoniae and Ceftaroline is a novel cephalosporin recently resistant Gram-positive infections that fail approved in children for treatment of acute first-line antimicrobial agents. However, bacterial skin and soft tissue infections and limited data are available on tolerability in community-acquired bacterial pneumonia neonates and infants younger than 2 months (CABP) caused by methicillin-resistant of age, and on pharmacokinetic characteristics Staphylococcus aureus, Streptococcus pneumoniae in children with chronic medical conditions and other susceptible bacteria. With a favorable and those with invasive, complicated tolerability profile and efficacy proven in infections. In this review, the microbiological pediatric patients and excellent in vitro profile of ceftaroline, its mechanism of action, activity against resistant Gram-positive and and pharmacokinetic profile will be presented. Gram-negative bacteria, ceftaroline may serve Additionally, clinical evidence for use in as a therapeutic option for polymicrobial pediatric patients and proposed place in therapy is discussed. Enhanced content To view enhanced content for this article go to http://www.medengine.com/Redeem/ 1F47F0601BB3F2DD. Keywords: Antibiotic resistance; Ceftaroline J. Yim (&) fosamil; Children; Methicillin-resistant St. John Hospital and Medical Center, Detroit, MI, Staphylococcus aureus; Streptococcus pneumoniae USA e-mail: [email protected] L. -

Pharmacodynamic Evaluation of Suppression of in Vitro Resistance In
www.nature.com/scientificreports OPEN Pharmacodynamic evaluation of suppression of in vitro resistance in Acinetobacter baumannii strains using polymyxin B‑based combination therapy Nayara Helisandra Fedrigo1, Danielle Rosani Shinohara1, Josmar Mazucheli2, Sheila Alexandra Belini Nishiyama1, Floristher Elaine Carrara‑Marroni3, Frederico Severino Martins4, Peijuan Zhu5, Mingming Yu6, Sherwin Kenneth B. Sy2 & Maria Cristina Bronharo Tognim1* The emergence of polymyxin resistance in Gram‑negative bacteria infections has motivated the use of combination therapy. This study determined the mutant selection window (MSW) of polymyxin B alone and in combination with meropenem and fosfomycin against A. baumannii strains belonging to clonal lineages I and III. To evaluate the inhibition of in vitro drug resistance, we investigate the MSW‑derived pharmacodynamic indices associated with resistance to polymyxin B administrated regimens as monotherapy and combination therapy, such as the percentage of each dosage interval that free plasma concentration was within the MSW (%TMSW) and the percentage of each dosage interval that free plasma concentration exceeded the mutant prevention concentration (%T>MPC). The MSW of polymyxin B varied between 1 and 16 µg/mL for polymyxin B‑susceptible strains. The triple combination of polymyxin B with meropenem and fosfomycin inhibited the polymyxin B‑resistant subpopulation in meropenem‑resistant isolates and polymyxin B plus meropenem as a double combination sufciently inhibited meropenem‑intermediate, and susceptible strains. T>MPC 90% was reached for polymyxin B in these combinations, while %TMSW was 0 against all strains. TMSW for meropenem and fosfomycin were also reduced. Efective antimicrobial combinations signifcantly reduced MSW. The MSW‑derived pharmacodynamic indices can be used for the selection of efective combination regimen to combat the polymyxin B‑resistant strain. -

General Items
Essential Medicines List (EML) 2019 Application for the inclusion of imipenem/cilastatin, meropenem and amoxicillin/clavulanic acid in the WHO Model List of Essential Medicines, as reserve second-line drugs for the treatment of multidrug-resistant tuberculosis (complementary lists of anti-tuberculosis drugs for use in adults and children) General items 1. Summary statement of the proposal for inclusion, change or deletion This application concerns the updating of the forthcoming WHO Model List of Essential Medicines (EML) and WHO Model List of Essential Medicines for Children (EMLc) to include the following medicines: 1) Imipenem/cilastatin (Imp-Cln) to the main list but NOT the children’s list (it is already mentioned on both lists as an option in section 6.2.1 Beta Lactam medicines) 2) Meropenem (Mpm) to both the main and the children’s lists (it is already on the list as treatment for meningitis in section 6.2.1 Beta Lactam medicines) 3) Clavulanic acid to both the main and the children’s lists (it is already listed as amoxicillin/clavulanic acid (Amx-Clv), the only commercially available preparation of clavulanic acid, in section 6.2.1 Beta Lactam medicines) This application makes reference to amendments recommended in particular to section 6.2.4 Antituberculosis medicines in the latest editions of both the main EML (20th list) and the EMLc (6th list) released in 2017 (1),(2). On the basis of the most recent Guideline Development Group advising WHO on the revision of its guidelines for the treatment of multidrug- or rifampicin-resistant (MDR/RR-TB)(3), the applicant considers that the three agents concerned be viewed as essential medicines for these forms of TB in countries. -

Severe Sepsis and Septic Shock Antibiotic Guide
Stanford Health Issue Date: 05/2017 Stanford Antimicrobial Safety and Sustainability Program Severe Sepsis and Septic Shock Antibiotic Guide Table 1: Antibiotic selection options for healthcare associated and/or immunocompromised patients • Healthcare associated: intravenous therapy, wound care, or intravenous chemotherapy within the prior 30 days, residence in a nursing home or other long-term care facility, hospitalization in an acute care hospital for two or more days within the prior 90 days, attendance at a hospital or hemodialysis clinic within the prior 30 days • Immunocompromised: Receiving chemotherapy, known systemic cancer not in remission, ANC <500, severe cell-mediated immune deficiency Table 2: Antibiotic selection options for community acquired, immunocompetent patients Table 3: Antibiotic selection options for patients with simple sepsis, community acquired, immunocompetent patients requiring hospitalization. Risk Factors for Select Organisms P. aeruginosa MRSA Invasive Candidiasis VRE (and other resistant GNR) Community acquired: • Known colonization with MDROs • Central venous catheter • Liver transplant • Prior IV antibiotics within 90 day • Recent MRSA infection • Broad-spectrum antibiotics • Known colonization • Known colonization with MDROs • Known MRSA colonization • + 1 of the following risk factors: • Prolonged broad antibacterial • Skin & Skin Structure and/or IV access site: ♦ Parenteral nutrition therapy Hospital acquired: ♦ Purulence ♦ Dialysis • Prolonged profound • Prior IV antibiotics within 90 days ♦ Abscess -
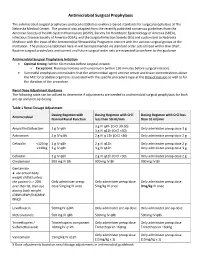
Antimicrobial Surgical Prophylaxis
Antimicrobial Surgical Prophylaxis The antimicrobial surgical prophylaxis protocol establishes evidence-based standards for surgical prophylaxis at The Nebraska Medical Center. The protocol was adapted from the recently published consensus guidelines from the American Society of Health-System Pharmacists (ASHP), Society for Healthcare Epidemiology of America (SHEA), Infectious Disease Society of America (IDSA), and the Surgical Infection Society (SIS) and customized to Nebraska Medicine with the input of the Antimicrobial Stewardship Program in concert with the various surgical groups at the institution. The protocol established here-in will be implemented via standard order sets utilized within One Chart. Routine surgical prophylaxis and current and future surgical order sets are expected to conform to this guidance. Antimicrobial Surgical Prophylaxis Initiation Optimal timing: Within 60 minutes before surgical incision o Exceptions: Fluoroquinolones and vancomycin (within 120 minutes before surgical incision) Successful prophylaxis necessitates that the antimicrobial agent achieve serum and tissue concentrations above the MIC for probable organisms associated with the specific procedure type at the time of incision as well as for the duration of the procedure. Renal Dose Adjustment Guidance The following table can be utilized to determine if adjustments are needed to antimicrobial surgical prophylaxis for both pre-op and post-op dosing. Table 1 Renal Dosage Adjustment Dosing Regimen with Dosing Regimen with CrCl Dosing Regimen with -

Zinforo, INN Ceftaroline Fosamil
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT Zinforo 600 mg powder for concentrate for solution for infusion 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each vial contains ceftaroline fosamil acetic acid solvate monohydrate equivalent to 600 mg ceftaroline fosamil. After reconstitution, 1 ml of the solution contains 30 mg of ceftaroline fosamil. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Powder for concentrate for solution for infusion. A pale yellowish-white to light yellow powder. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Zinforo is indicated in adults for the treatment of the following infections (see sections 4.4 and 5.1): • Complicated skin and soft tissue infections (cSSTI) • Community-acquired pneumonia (CAP) Consideration should be given to official guidance on the appropriate use of antibacterial agents. 4.2 Posology and method of administration Posology For the treatment of cSSTI and CAP, the recommended dose is 600 mg administered every 12 hours by intravenous infusion over 60 minutes in patients aged 18 years or older. The recommended treatment duration for cSSTI is 5 to 14 days and the recommended duration of treatment for CAP is 5 to 7 days. Special populations Elderly patients (≥ 65 years) No dosage adjustment is required for the elderly with creatinine clearance values > 50 ml/min (see section 5.2). Renal impairment The dose should be adjusted when creatinine clearance (CrCL) is ≤ 50 ml/min, as shown below (see sections 4.4 and 5.2). Creatinine clearance Dosage regimen Frequency (ml/min) > 30 to ≤ 50 400 mg intravenously (over 60 minutes) every 12 hours 2 There is insufficient data to make specific dosage adjustment recommendations for patients with severe renal impairment (CrCL ≤ 30 ml/min) and end-stage renal disease (ESRD), including patients undergoing haemodialysis (see section 4.4). -

Summary of Product Characteristics
SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT Augmentin 125 mg/31.25 mg/5 ml powder for oral suspension Augmentin 250 mg/62.5 mg/5 ml powder for oral suspension 2. QUALITATIVE AND QUANTITATIVE COMPOSITION When reconstituted, every ml of oral suspension contains amoxicillin trihydrate equivalent to 25 mg amoxicillin and potassium clavulanate equivalent to 6.25 mg of clavulanic acid. Excipients with known effect Every ml of oral suspension contains 2.5 mg aspartame (E951). The flavouring in Augmentin contains maltodextrin (glucose) (see section 4.4). This medicine contains less than 1 mmol sodium (23 mg) per ml, that is to say essentially ‘sodium- free’. When reconstituted, every ml of oral suspension contains amoxicillin trihydrate equivalent to 50 mg amoxicillin and potassium clavulanate equivalent to 12.5 mg of clavulanic acid. Excipients with known effect Every ml of oral suspension contains 2.5 mg aspartame (E951). The flavouring in Augmentin contains maltodextrin (glucose) (see section 4.4). This medicine contains less than 1 mmol sodium (23 mg) per ml, that is to say essentially ‘sodium- free’. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Powder for oral suspension. Off-white powder. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Augmentin is indicated for the treatment of the following infections in adults and children (see sections 4.2, 4.4 and 5.1): • Acute bacterial sinusitis (adequately diagnosed) • Acute otitis media • Acute exacerbations of chronic bronchitis (adequately diagnosed) • Community acquired pneumonia • Cystitis • Pyelonephritis 2 • Skin and soft tissue infections in particular cellulitis, animal bites, severe dental abscess with spreading cellulitis • Bone and joint infections, in particular osteomyelitis. -

Australian Public Assessment Refport for Ceftaroline Fosamil (Zinforo)
Australian Public Assessment Report for ceftaroline fosamil Proprietary Product Name: Zinforo Sponsor: AstraZeneca Pty Ltd May 2013 Therapeutic Goods Administration About the Therapeutic Goods Administration (TGA) • The Therapeutic Goods Administration (TGA) is part of the Australian Government Department of Health and Ageing, and is responsible for regulating medicines and medical devices. • The TGA administers the Therapeutic Goods Act 1989 (the Act), applying a risk management approach designed to ensure therapeutic goods supplied in Australia meet acceptable standards of quality, safety and efficacy (performance), when necessary. • The work of the TGA is based on applying scientific and clinical expertise to decision- making, to ensure that the benefits to consumers outweigh any risks associated with the use of medicines and medical devices. • The TGA relies on the public, healthcare professionals and industry to report problems with medicines or medical devices. TGA investigates reports received by it to determine any necessary regulatory action. • To report a problem with a medicine or medical device, please see the information on the TGA website <http://www.tga.gov.au>. About AusPARs • An Australian Public Assessment Record (AusPAR) provides information about the evaluation of a prescription medicine and the considerations that led the TGA to approve or not approve a prescription medicine submission. • AusPARs are prepared and published by the TGA. • An AusPAR is prepared for submissions that relate to new chemical entities, generic medicines, major variations, and extensions of indications. • An AusPAR is a static document, in that it will provide information that relates to a submission at a particular point in time. • A new AusPAR will be developed to reflect changes to indications and/or major variations to a prescription medicine subject to evaluation by the TGA. -

Penicillin Allergy Guidance Document
Penicillin Allergy Guidance Document Key Points Background Careful evaluation of antibiotic allergy and prior tolerance history is essential to providing optimal treatment The true incidence of penicillin hypersensitivity amongst patients in the United States is less than 1% Alterations in antibiotic prescribing due to reported penicillin allergy has been shown to result in higher costs, increased risk of antibiotic resistance, and worse patient outcomes Cross-reactivity between truly penicillin allergic patients and later generation cephalosporins and/or carbapenems is rare Evaluation of Penicillin Allergy Obtain a detailed history of allergic reaction Classify the type and severity of the reaction paying particular attention to any IgE-mediated reactions (e.g., anaphylaxis, hives, angioedema, etc.) (Table 1) Evaluate prior tolerance of beta-lactam antibiotics utilizing patient interview or the electronic medical record Recommendations for Challenging Penicillin Allergic Patients See Figure 1 Follow-Up Document tolerance or intolerance in the patient’s allergy history Consider referring to allergy clinic for skin testing Created July 2017 by Macey Wolfe, PharmD; John Schoen, PharmD, BCPS; Scott Bergman, PharmD, BCPS; Sara May, MD; and Trevor Van Schooneveld, MD, FACP Disclaimer: This resource is intended for non-commercial educational and quality improvement purposes. Outside entities may utilize for these purposes, but must acknowledge the source. The guidance is intended to assist practitioners in managing a clinical situation but is not mandatory. The interprofessional group of authors have made considerable efforts to ensure the information upon which they are based is accurate and up to date. Any treatments have some inherent risk. Recommendations are meant to improve quality of patient care yet should not replace clinical judgment. -

DICLOXACILLIN MYLAN Dicloxacillin Sodium Capsules
AUSTRALIAN PRODUCT INFORMATION DICLOXACILLIN MYLAN Dicloxacillin sodium capsules 1 NAME OF THE MEDICINE Dicloxacillin (as dicloxacillin sodium) 2 QUALITATIVE AND QUANTITATIVE COMPOSITION Each capsule contains dicloxacillin sodium equivalent to 250 mg or 500 mg dicloxacillin as the active ingredient. For the full list of excipients, see Section 6.1 LIST OF EXCIPIENTS. 3 PHARMACEUTICAL FORM DICLOXACILLIN : Dicloxacillin 250 mg capsule: Size 2 capsule with white opaque body and cap, MYLAN 250 marked ‘DX’ on the cap and ‘250’ on the body in black DICLOXACILLIN : Dicloxacillin 500 mg capsule: Size 0 capsule with white opaque body and cap, MYLAN 500 marked ‘DX’ on the cap and ‘500’ on the body in black 4 CLINICAL PARTICULARS 4.1 THERAPEUTIC INDICATIONS Treatment of confirmed or suspected staphylococcal and other Gram positive coccal infections, including skin and skin structure and wound infections, infected burns, cellulitis, osteomyelitis and pneumonia (note: benzylpenicillin is the drug of choice for the treatment of streptococcal pneumonia). Bacteriological studies should be performed to determine the causative organisms and their susceptibility to dicloxacillin. Dicloxacillin has less intrinsic antibacterial activity and a narrower spectrum than benzylpenicillin. Dicloxacillin should therefore not be used in infections due to organisms susceptible to benzylpenicillin. Important Note: When it is judged necessary that treatment is initiated before definitive culture and sensitivity results are known, if the microbiology report later indicates that the infection is due to an organism other than a benzylpenicillin resistant staphylococcus sensitive to dicloxacillin, the physician is advised to continue therapy with a drug other than dicloxacillin or any other penicillinase-resistant penicillin. 4.2 DOSE AND METHOD OF ADMINISTRATION Microbiological studies to determine the causative organism and their susceptibility to the penicillinase resistant penicillins should be performed. -

Approximately 15 Units of Penicillinase Were Preincubated with 4 Ml of Phenylisoxazolyl Penicillins (5 Mg/Ml) at 30°C
INACTIVATION OF STAPHYLOCOCCAL PENICILLINASE BY DICLOXACILLIN Hitoshi Sagai and Tetsu Saito Research Laboratories, Toyo Jozo Co., Ltd. Ohito-cho, Shizuoka-ken, 410-23, Japan (Received for publication February 12, 1973) Staphylococcal penicillinase was inactivated by treatment with a relatively low concentration of methyldichlorophenyl-isoxazolyl penicillin (dicloxacillin). Inactivated enzyme was isolated by gel-filtration and reactivated by incubation at 37°C. It is suggested that the inactivated enzyme is penicilloyl enzyme which is readily hydrolyzed to active enzyme. The rates of hydrolysis of /3-lactamase-sensitive penicillins (such as benzylpenicillin and aminobenzylpenicillin) by penicillinase were greatly decreased by the addition of /3-lactamase resistant penicillins1'23. Gourevitch and his coworkers reported that the inactivation of cell-bound staphylococcal penicillinase occurred when the enzyme was preincubated with dimethoxyphenyl penicillin (methicillin), and the amount of inactivated enzyme corresponded to the amount of hydrolyzed methicillin3). On the other hand, Richmond demonstrated that the purified exo-enzyme of staphylococci degraded more than 85% of added methicillin without inactivation of the enzyme4). In this paper, we deal with the inactivation of the extracellular staphylococcal enzyme by dicloxacillin and propose a tentative mechanism to account for the inactivation. Materials and Methods Drugs : Benzylpenicillin potassium.salt, aminobenzylpenicillin (ampicillin) sodium salt, methyl- chlorophenylisoxazolyl penicillin (cloxacillin) sodium salt and dicloxacillin sodium salt (Toyo Jozo Co., Ltd.) were used. Organism: Staphylococcus aureus 0003 was used as the penicillinase source. The strain was of clincal origin and its penicillinase was inducible. Preparation of penicillinase: Partially purified enzyme was prepared according to the method reported by Richmond43. The supernatant fluid of methicillin-induced S.