Australian Public Assessment Refport for Ceftaroline Fosamil (Zinforo)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dabigatran Amoxicillin Clavulanate IV Treatment in the Community
BEST PRACTICE 38 SEPTEMBER 2011 Dabigatran Amoxicillin clavulanate bpac nz IV treatment in the community better medicin e Editor-in-chief We would like to acknowledge the following people for Professor Murray Tilyard their guidance and expertise in developing this edition: Professor Carl Burgess, Wellington Editor Dr Gerry Devlin, Hamilton Rebecca Harris Dr John Fink, Christchurch Dr Lisa Houghton, Dunedin Programme Development Dr Rosemary Ikram, Christchurch Mark Caswell Dr Sisira Jayathissa, Wellington Rachael Clarke Kate Laidlow, Rotorua Peter Ellison Dr Hywel Lloyd, GP Reviewer, Dunedin Julie Knight Associate Professor Stewart Mann, Wellington Noni Richards Dr Richard Medlicott, Wellington Dr AnneMarie Tangney Dr Alan Panting, Nelson Dr Sharyn Willis Dr Helen Patterson, Dunedin Dave Woods David Rankin, Wellington Report Development Dr Ralph Stewart, Auckland Justine Broadley Dr Neil Whittaker, GP Reviewer, Nelson Tim Powell Dr Howard Wilson, Akaroa Design Michael Crawford Best Practice Journal (BPJ) ISSN 1177-5645 Web BPJ, Issue 38, September 2011 Gordon Smith BPJ is published and owned by bpacnz Ltd Management and Administration Level 8, 10 George Street, Dunedin, New Zealand. Jaala Baldwin Bpacnz Ltd is an independent organisation that promotes health Kaye Baldwin care interventions which meet patients’ needs and are evidence Tony Fraser based, cost effective and suitable for the New Zealand context. Kyla Letman We develop and distribute evidence based resources which describe, facilitate and help overcome the barriers to best Clinical Advisory Group practice. Clive Cannons nz Michele Cray Bpac Ltd is currently funded through contracts with PHARMAC and DHBNZ. Margaret Gibbs nz Dr Rosemary Ikram Bpac Ltd has five shareholders: Procare Health, South Link Health, General Practice NZ, the University of Otago and Pegasus Dr Cam Kyle Health. -

Cusumano-Et-Al-2017.Pdf
International Journal of Infectious Diseases 63 (2017) 1–6 Contents lists available at ScienceDirect International Journal of Infectious Diseases journal homepage: www.elsevier.com/locate/ijid Rapidly growing Mycobacterium infections after cosmetic surgery in medical tourists: the Bronx experience and a review of the literature a a b c b Lucas R. Cusumano , Vivy Tran , Aileen Tlamsa , Philip Chung , Robert Grossberg , b b, Gregory Weston , Uzma N. Sarwar * a Albert Einstein College of Medicine, Montefiore Medical Center, Bronx, New York, USA b Division of Infectious Diseases, Department of Medicine, Albert Einstein College of Medicine, Montefiore Medical Center, Bronx, New York, USA c Department of Pharmacy, Nebraska Medicine, Omaha, Nebraska, USA A R T I C L E I N F O A B S T R A C T Article history: Background: Medical tourism is increasingly popular for elective cosmetic surgical procedures. However, Received 10 May 2017 medical tourism has been accompanied by reports of post-surgical infections due to rapidly growing Received in revised form 22 July 2017 mycobacteria (RGM). The authors’ experience working with patients with RGM infections who have Accepted 26 July 2017 returned to the USA after traveling abroad for cosmetic surgical procedures is described here. Corresponding Editor: Eskild Petersen, Methods: Patients who developed RGM infections after undergoing cosmetic surgeries abroad and who ?Aarhus, Denmark presented at the Montefiore Medical Center (Bronx, New York, USA) between August 2015 and June 2016 were identified. A review of patient medical records was performed. Keywords: Results: Four patients who presented with culture-proven RGM infections at the sites of recent cosmetic Mycobacterium abscessus complex procedures were identified. -

Flucloxacillin Capsules in This Leaflet: 1 What Flucloxacillin Capsules Are
Flucloxacillin capsules This information is a summary only. It does not contain all information about this medicine. If you would like more information about the medicine you are taking, check with your doctor or other health care provider. No rights can be derived from the information provided in this medicine leaflet. In this leaflet: If you take more than you should 1 What Flucloxacillin capsules are and what they are used for If you (or someone else) swallow a lot of capsules at the same time, or you think a 2 Before you take child may have swallowed any contact your nearest hospital casualty department 3 How to take or tell your doctor immediately. Symptoms of an overdose include feeling or 4 Possible side effects being sick and diarrhoea. 5 How to store If you forget to take the capsules 1 What Flucloxacillin capsules are and what they are used for Do not take a double dose to make up for a forgotten dose. If you forget to take a Flucloxacillin is an antibiotic used to treat infections by killing the bacteria that dose take it as soon as you remember it and carry on as before, try to wait about can cause them. It belongs to a group of antibiotics called “penicillins”. four hours before taking the next dose. Flucloxacillin capsules are used to treat: • chest infections If you stop taking the capsules • throat or nose infections Do not stop treatment early because some bacteria may survive and cause the • ear infections infection to come back. • skin and soft tissue infections • heart infections 4 Possible side effects • bones and joints infections Like all medicines, Flucloxacillin capsules can cause side effects, although not • meningitis everybody gets them. -

Clinical Management of Staphylococcus Aureus Bacteremia in Neonates, Children, and Adolescents
Clinical Management of Staphylococcus aureus Bacteremia in Neonates, Children, and Adolescents Brendan J. McMullan, BMed (Hons), FRACP, FRCPA,a,b,c,* Anita J. Campbell, MBBS, DCH, DipPID, FRACP,d,e,f,* Christopher C. Blyth, MBBS (Hons), PhD, DCH, FRACP, FRCPA,d,e,f,g J. Chase McNeil, BS, MD,h Christopher P. Montgomery, BA, MD,i,j Steven Y.C. Tong, MBBS (Hons), PhD, FRACP,k,l Asha C. Bowen, BA, MBBS, PhD, FRACPd,e,f,k,m Staphylococcus aureus is a common cause of community and health abstract – care associated bacteremia, with authors of recent studies estimating the aDepartment of Immunology and Infectious Diseases, incidence of S aureus bacteremia (SAB) in high-income countries between 8 Sydney Children’s Hospital, Randwick, New South Wales, , Australia; bSchool of Women’s and Children’s Health, and 26 per 100 000 children per year. Despite this, 300 children worldwide University of New South Wales, Sydney, New South Wales, have ever been randomly assigned into clinical trials to assess the efficacy Australia; cNational Centre for Infections in Cancer, of treatment of SAB. A panel of infectious diseases physicians with clinical University of Melbourne, Melbourne, Victoria, Australia; dDepartment of Infectious Diseases, Perth Children’s and research interests in pediatric SAB identified 7 key clinical questions. Hospital, Nedlands, Western Australia, Australia; The available literature is systematically appraised, summarizing SAB eWesfarmers Centre of Vaccines and Infectious Diseases, Telethon Kids Institute, Nedlands, Western Australia, -
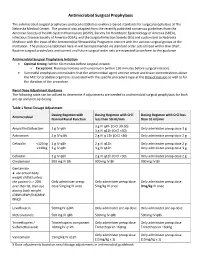
Antimicrobial Surgical Prophylaxis
Antimicrobial Surgical Prophylaxis The antimicrobial surgical prophylaxis protocol establishes evidence-based standards for surgical prophylaxis at The Nebraska Medical Center. The protocol was adapted from the recently published consensus guidelines from the American Society of Health-System Pharmacists (ASHP), Society for Healthcare Epidemiology of America (SHEA), Infectious Disease Society of America (IDSA), and the Surgical Infection Society (SIS) and customized to Nebraska Medicine with the input of the Antimicrobial Stewardship Program in concert with the various surgical groups at the institution. The protocol established here-in will be implemented via standard order sets utilized within One Chart. Routine surgical prophylaxis and current and future surgical order sets are expected to conform to this guidance. Antimicrobial Surgical Prophylaxis Initiation Optimal timing: Within 60 minutes before surgical incision o Exceptions: Fluoroquinolones and vancomycin (within 120 minutes before surgical incision) Successful prophylaxis necessitates that the antimicrobial agent achieve serum and tissue concentrations above the MIC for probable organisms associated with the specific procedure type at the time of incision as well as for the duration of the procedure. Renal Dose Adjustment Guidance The following table can be utilized to determine if adjustments are needed to antimicrobial surgical prophylaxis for both pre-op and post-op dosing. Table 1 Renal Dosage Adjustment Dosing Regimen with Dosing Regimen with CrCl Dosing Regimen with -

Antimicrobial Stewardship Guidance
Antimicrobial Stewardship Guidance Federal Bureau of Prisons Clinical Practice Guidelines March 2013 Clinical guidelines are made available to the public for informational purposes only. The Federal Bureau of Prisons (BOP) does not warrant these guidelines for any other purpose, and assumes no responsibility for any injury or damage resulting from the reliance thereof. Proper medical practice necessitates that all cases are evaluated on an individual basis and that treatment decisions are patient-specific. Consult the BOP Clinical Practice Guidelines Web page to determine the date of the most recent update to this document: http://www.bop.gov/news/medresources.jsp Federal Bureau of Prisons Antimicrobial Stewardship Guidance Clinical Practice Guidelines March 2013 Table of Contents 1. Purpose ............................................................................................................................................. 3 2. Introduction ...................................................................................................................................... 3 3. Antimicrobial Stewardship in the BOP............................................................................................ 4 4. General Guidance for Diagnosis and Identifying Infection ............................................................. 5 Diagnosis of Specific Infections ........................................................................................................ 6 Upper Respiratory Infections (not otherwise specified) .............................................................................. -

Susceptibility of Pseudomonas Cepacia Isolated from Children with Cystic Fibrosis1
003 1-3998/86/2011-1174$02.00/0 PEDIATRIC RESEARCH Vol. 20, No. 1 1, 1986 Copyright O 1986 International Pediatric Research Foundation, Inc. Printed in (I.S.A. Decreased Baseline P-Lactamase Production and Inducibility associated with Increased Piperacillin Susceptibility of Pseudomonas cepacia Isolated from Children with Cystic Fibrosis1 CLAUDIO CHIESA,~PAULINE H. LABROZZI, AND STEPHEN C. ARONOFF Department of Pediatrics [C.C., P.H.L., S.C.A.], Case- Western Reserve University School ofMedicine and the Division of Pediatric Infectious Diseases [S.C.A.], Rainbow Babies and Children's Hospital, Cleveland, Ohio ABSTRACT. The incidence of pulmonary infections in creased since 1978 and is now recovered from 20% of patients children with cystic fibrosis caused by Pseudomonas ce- in some centers (3, 4). pacia, an organism which may possess an inducible 8- Sputum isolates of P. cepacia from children with cystic fibrosis lactamase, has increased since 1978. Seven of 13 sputum are resistant to most P-lactam agents in vitro. In an in vitro study isolates of P. cepacia from children with cystic fibrosis comparing the susceptibilities of 62 consecutive sputum isolates, were classified as inducible by quantitative enzyme produc- concentrations of 64 pg/ml of aztreonam and piperacillin were tion following preincubation with 100, 200, or 400 pg/ml required to inhibit 79 and 87.1 % of the bacterial population, of cefoxitin. The recovery of inducible strains tended to be respectively; 90% of the isolates were inhibited by 8 pg/ml of associated with recent ceftazidime therapy. Susceptibility ceftazidime (5). In vitro, ceftazidime has proven more active to aztreonam, ceftazidime, and piperacillin alone or com- against P. -

Zinforo, INN Ceftaroline Fosamil
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT Zinforo 600 mg powder for concentrate for solution for infusion 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each vial contains ceftaroline fosamil acetic acid solvate monohydrate equivalent to 600 mg ceftaroline fosamil. After reconstitution, 1 ml of the solution contains 30 mg of ceftaroline fosamil. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Powder for concentrate for solution for infusion. A pale yellowish-white to light yellow powder. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Zinforo is indicated in adults for the treatment of the following infections (see sections 4.4 and 5.1): • Complicated skin and soft tissue infections (cSSTI) • Community-acquired pneumonia (CAP) Consideration should be given to official guidance on the appropriate use of antibacterial agents. 4.2 Posology and method of administration Posology For the treatment of cSSTI and CAP, the recommended dose is 600 mg administered every 12 hours by intravenous infusion over 60 minutes in patients aged 18 years or older. The recommended treatment duration for cSSTI is 5 to 14 days and the recommended duration of treatment for CAP is 5 to 7 days. Special populations Elderly patients (≥ 65 years) No dosage adjustment is required for the elderly with creatinine clearance values > 50 ml/min (see section 5.2). Renal impairment The dose should be adjusted when creatinine clearance (CrCL) is ≤ 50 ml/min, as shown below (see sections 4.4 and 5.2). Creatinine clearance Dosage regimen Frequency (ml/min) > 30 to ≤ 50 400 mg intravenously (over 60 minutes) every 12 hours 2 There is insufficient data to make specific dosage adjustment recommendations for patients with severe renal impairment (CrCL ≤ 30 ml/min) and end-stage renal disease (ESRD), including patients undergoing haemodialysis (see section 4.4). -

Summary of Product Characteristics
SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT Augmentin 125 mg/31.25 mg/5 ml powder for oral suspension Augmentin 250 mg/62.5 mg/5 ml powder for oral suspension 2. QUALITATIVE AND QUANTITATIVE COMPOSITION When reconstituted, every ml of oral suspension contains amoxicillin trihydrate equivalent to 25 mg amoxicillin and potassium clavulanate equivalent to 6.25 mg of clavulanic acid. Excipients with known effect Every ml of oral suspension contains 2.5 mg aspartame (E951). The flavouring in Augmentin contains maltodextrin (glucose) (see section 4.4). This medicine contains less than 1 mmol sodium (23 mg) per ml, that is to say essentially ‘sodium- free’. When reconstituted, every ml of oral suspension contains amoxicillin trihydrate equivalent to 50 mg amoxicillin and potassium clavulanate equivalent to 12.5 mg of clavulanic acid. Excipients with known effect Every ml of oral suspension contains 2.5 mg aspartame (E951). The flavouring in Augmentin contains maltodextrin (glucose) (see section 4.4). This medicine contains less than 1 mmol sodium (23 mg) per ml, that is to say essentially ‘sodium- free’. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Powder for oral suspension. Off-white powder. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Augmentin is indicated for the treatment of the following infections in adults and children (see sections 4.2, 4.4 and 5.1): • Acute bacterial sinusitis (adequately diagnosed) • Acute otitis media • Acute exacerbations of chronic bronchitis (adequately diagnosed) • Community acquired pneumonia • Cystitis • Pyelonephritis 2 • Skin and soft tissue infections in particular cellulitis, animal bites, severe dental abscess with spreading cellulitis • Bone and joint infections, in particular osteomyelitis. -

Comparative Ceftaroline Activity Tested Against Staphylococcus Aureus Associated with Acute Bacterial Skin and Skin Structure Infection from a Tertiary Care Center
RESEARCH PAPER Medical Science Volume : 5 | Issue : 4 | April 2015 | ISSN - 2249-555X Comparative Ceftaroline Activity Tested Against Staphylococcus Aureus Associated with Acute Bacterial Skin and Skin Structure Infection from A Tertiary Care Center KEYWORDS MRSA, Ceftaroline, vancomycin Dr. Umamageswari S. S. M. Dr. S. Habeeb Mohammed Dr. Shameem Banu Associate Professor- Microbiology Associate Professor-Surgery Professor and HOD- Microbiology Chettinad Hospital & Research Chettinad Hospital & Research Chettinad Hospital & Research Insitute Insitute Insitute Dr. Jayanthi S. Associate Professor- Microbiology Chettinad Hospital & Research Insitute ABSTRACT Methicillin resistant Staphylococcus aureus (MRSA), commonest cause of acute bacterial skin and skin structure infection (ABSSSI) has only limited treatment options like vancomycin, Linezolid and teicoplanin. Staphylococcus aureus with reduced susceptibilities to vancomycin is developing nowadays among nosocomial infec- tion. A newest cephalosporin – Ceftaroline has received FDA approval for the treatment of ABSSSI recently has unique activity against MRSA. The aim of the study is to know the prevalence of Staphylococcus aureus in ABSSSI patients and to evaluate the in vitro activity of Ceftaroline, in comparison with other drugs. Minimum Inhibitory Concentration for vancomycin and Ceftaroline were determined by E-test strips. Out of 235 Staphylococcus aureus isolated from ABSSSI patient 32% were MRSA. No vancomycin resistant strain isolated in our study. In vitro, Ceftaroline control the growth of MRSA very effectively at 2mg/ml itself. Ceftaroline is a most welcomed drug in the treatment of MRSA. INTRODUCTION: MATERIALS AND METHODS: Complicated skin and skin structure infection (CSSSIs), now This study was carried out in Chettinad health city and known by the new US FDA as Acute bacterial skin and skin Research Institute, a tertiary care center hospital (Chen- structure infections (ABSSSI) are the most common infec- nai) between April 2013 and May 2014. -

Penicillin Allergy Guidance Document
Penicillin Allergy Guidance Document Key Points Background Careful evaluation of antibiotic allergy and prior tolerance history is essential to providing optimal treatment The true incidence of penicillin hypersensitivity amongst patients in the United States is less than 1% Alterations in antibiotic prescribing due to reported penicillin allergy has been shown to result in higher costs, increased risk of antibiotic resistance, and worse patient outcomes Cross-reactivity between truly penicillin allergic patients and later generation cephalosporins and/or carbapenems is rare Evaluation of Penicillin Allergy Obtain a detailed history of allergic reaction Classify the type and severity of the reaction paying particular attention to any IgE-mediated reactions (e.g., anaphylaxis, hives, angioedema, etc.) (Table 1) Evaluate prior tolerance of beta-lactam antibiotics utilizing patient interview or the electronic medical record Recommendations for Challenging Penicillin Allergic Patients See Figure 1 Follow-Up Document tolerance or intolerance in the patient’s allergy history Consider referring to allergy clinic for skin testing Created July 2017 by Macey Wolfe, PharmD; John Schoen, PharmD, BCPS; Scott Bergman, PharmD, BCPS; Sara May, MD; and Trevor Van Schooneveld, MD, FACP Disclaimer: This resource is intended for non-commercial educational and quality improvement purposes. Outside entities may utilize for these purposes, but must acknowledge the source. The guidance is intended to assist practitioners in managing a clinical situation but is not mandatory. The interprofessional group of authors have made considerable efforts to ensure the information upon which they are based is accurate and up to date. Any treatments have some inherent risk. Recommendations are meant to improve quality of patient care yet should not replace clinical judgment. -

DICLOXACILLIN MYLAN Dicloxacillin Sodium Capsules
AUSTRALIAN PRODUCT INFORMATION DICLOXACILLIN MYLAN Dicloxacillin sodium capsules 1 NAME OF THE MEDICINE Dicloxacillin (as dicloxacillin sodium) 2 QUALITATIVE AND QUANTITATIVE COMPOSITION Each capsule contains dicloxacillin sodium equivalent to 250 mg or 500 mg dicloxacillin as the active ingredient. For the full list of excipients, see Section 6.1 LIST OF EXCIPIENTS. 3 PHARMACEUTICAL FORM DICLOXACILLIN : Dicloxacillin 250 mg capsule: Size 2 capsule with white opaque body and cap, MYLAN 250 marked ‘DX’ on the cap and ‘250’ on the body in black DICLOXACILLIN : Dicloxacillin 500 mg capsule: Size 0 capsule with white opaque body and cap, MYLAN 500 marked ‘DX’ on the cap and ‘500’ on the body in black 4 CLINICAL PARTICULARS 4.1 THERAPEUTIC INDICATIONS Treatment of confirmed or suspected staphylococcal and other Gram positive coccal infections, including skin and skin structure and wound infections, infected burns, cellulitis, osteomyelitis and pneumonia (note: benzylpenicillin is the drug of choice for the treatment of streptococcal pneumonia). Bacteriological studies should be performed to determine the causative organisms and their susceptibility to dicloxacillin. Dicloxacillin has less intrinsic antibacterial activity and a narrower spectrum than benzylpenicillin. Dicloxacillin should therefore not be used in infections due to organisms susceptible to benzylpenicillin. Important Note: When it is judged necessary that treatment is initiated before definitive culture and sensitivity results are known, if the microbiology report later indicates that the infection is due to an organism other than a benzylpenicillin resistant staphylococcus sensitive to dicloxacillin, the physician is advised to continue therapy with a drug other than dicloxacillin or any other penicillinase-resistant penicillin. 4.2 DOSE AND METHOD OF ADMINISTRATION Microbiological studies to determine the causative organism and their susceptibility to the penicillinase resistant penicillins should be performed.