A Molecular Network of the Aging Brain Implicates INPPL1 and PLXNB1 in Alzheimer’S Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PLXNB1 (Plexin
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Gene Section Mini Review PLXNB1 (plexin B1) José Javier Gómez-Román, Montserrat Nicolas Martínez, Servando Lazuén Fernández, José Fernando Val-Bernal Department of Anatomical Pathology, Marques de Valdecilla University Hospital, Medical Faculty, University of Cantabria, Santander, Spain (JJGR, MN, SL, JFVB) Published in Atlas Database: March 2009 Online updated version: http://AtlasGeneticsOncology.org/Genes/PLXNB1ID43413ch3p21.html DOI: 10.4267/2042/44702 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2010 Atlas of Genetics and Cytogenetics in Oncology and Haematology Identity Pseudogene No. Other names: KIAA0407; MGC149167; OTTHUMP00000164806; PLEXIN-B1; PLXN5; SEP Protein HGNC (Hugo): PLXNB1 Location: 3p21.31 Description Local order: The Plexin B1 gene is located between 2135 Amino acids (AA). Plexins are receptors for axon FBXW12 and CCDC51 genes. molecular guidance molecules semaphorins. Plexin signalling is important in pathfinding and patterning of both neurons and developing blood vessels. Plexin-B1 is a surface cell receptor. When it binds to its ligand SEMA4D it activates several pathways by binding of cytoplasmic ligands, like RHOA activation and subsequent changes of the actin cytoskeleton, axon guidance, invasive growth and cell migration. It monomers and heterodimers with PLXNB2 after proteolytic processing. Binds RAC1 that has been activated by GTP binding. It binds PLXNA1 and by similarity ARHGEF11, Note ARHGEF12, ERBB2, MET, MST1R, RND1, NRP1 Size: 26,200 bases. and NRP2. Orientation: minus strand. This family features the C-terminal regions of various plexins. The cytoplasmic region, which has been called DNA/RNA a SEX domain in some members of this family is involved in downstream signalling pathways, by Description interaction with proteins such as Rac1, RhoD, Rnd1 and other plexins. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Microrna Expression Signature of Oral Squamous Cell Carcinoma: Functional Role of Microrna-26A&Sol;B in the Modulation of No
FULL PAPER British Journal of Cancer (2015) 112, 891–900 | doi: 10.1038/bjc.2015.19 Keywords: microRNA; oral squamous cell carcinoma; miR-26a; miR-26b; tumour suppressor; TMEM184B; expression signature MicroRNA expression signature of oral squamous cell carcinoma: functional role of microRNA-26a/b in the modulation of novel cancer pathways I Fukumoto1,2, T Hanazawa2, T Kinoshita1,2, N Kikkawa2, K Koshizuka1,2, Y Goto1, R Nishikawa1, T Chiyomaru3, H Enokida3, M Nakagawa3, Y Okamoto2 and N Seki*,1 1Department of Functional Genomics, Chiba University Graduate School of Medicine, Chiba, Japan; 2Department of Otorhinolaryngology/Head and Neck Surgery, Chiba University Graduate School of Medicine, Chiba, Japan and 3Department of Urology, Graduate School of Medical and Dental Sciences, Kagoshima University, Kagoshima, Japan Background: MicroRNAs (miRNAs) have been shown to play major roles in carcinogenesis in a variety of cancers. The aim of this study was to determine the miRNA expression signature of oral squamous cell carcinoma (OSCC) and to investigate the functional roles of miR-26a and miR-26b in OSCC cells. Methods: An OSCC miRNA signature was constructed by PCR-based array methods. Functional studies of differentially expressed miRNAs were performed to investigate cell proliferation, migration, and invasion in OSCC cells. In silico database and genome-wide gene expression analyses were performed to identify molecular targets and pathways mediated by miR-26a/b. Results: miR-26a and miR-26b were significantly downregulated in OSCC. Restoration of both miR-26a and miR-26b in cancer cell lines revealed that these miRNAs significantly inhibited cancer cell migration and invasion. Our data demonstrated that the novel transmembrane TMEM184B gene was a direct target of miR-26a/b regulation. -

WO 2014/135655 Al 12 September 2014 (12.09.2014) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2014/135655 Al 12 September 2014 (12.09.2014) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C12Q 1/68 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/EP2014/054384 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 6 March 2014 (06.03.2014) KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, (25) Filing Language: English OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (26) Publication Language: English SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, (30) Priority Data: ZW. 13305253.0 6 March 2013 (06.03.2013) EP (84) Designated States (unless otherwise indicated, for every (71) Applicants: INSTITUT CURIE [FR/FR]; 26 rue d'Ulm, kind of regional protection available): ARIPO (BW, GH, F-75248 Paris cedex 05 (FR). CENTRE NATIONAL DE GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, LA RECHERCHE SCIENTIFIQUE [FR/FR]; 3 rue UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, Michel Ange, F-75016 Paris (FR). -
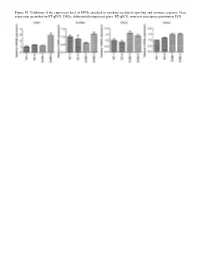
Figure S1. Validation of the Expression Level of Degs Enriched in Cytokine‑Mediated Signaling and Immune Response
Figure S1. Validation of the expression level of DEGs enriched in cytokine‑mediated signaling and immune response. Gene expression quantified by RT‑qPCR. DEGs, differentially expressed genes; RT‑qPCR, reverse‑transcription quantitative PCR. Figure S2. Validation of STAU1‑regulated AS events. IGV‑Sashimi plot revealed (A‑C) three A5SS AS events in three different genes. Reads distribution of each AS event was plotted in the left panel with the transcripts of each gene shown below. The sche‑ matic diagrams depict the structures of two AS events, AS1 (purple line) and AS2 (green line). The exon sequences are denoted by black boxes, the intron sequences by a horizontal line (right panel). RNA‑seq quantification and RT‑qPCR validation of ASEs are presented in the panels on the right. STAU1, double‑stranded RNA‑binding protein Staufen homolog 1; AS, alternative splicing; A5SS, alternative 5'splice site; RNA‑seq, RNA sequencing; RT‑qPCR, reverse‑transcription quantitative PCR. Error bars represent mean ± SEM. *P<0.05. Table SI. Primers used in gene validation experiments. IFIT2‑F CAGCCTACGGCAACTAAA IFIT2‑R GAGCCTTCTCAAAGCACA IFIT3‑F ACACCAAACAATGGCTAC IFIT3‑R TGGACAAACCCTCTAAAC OASL‑F AATGGTGACCGTGATGGG OASL‑R ACCTGAGGATGGAGCAGAG IFI27‑F TTCACTGCGGCGGGAATC IFI27‑R TGGCTGCTATGGAGGACGAG S1PR4‑F TGCTGAAGACGGTGCTGATG S1PR4‑R TGCGGAAGGAGTAGATGATGG CCL5‑F ACGACTGCTGGGTTGGAG CCL5‑R ACCCTGCTGCTTTGCCTA CCL2‑F CTAACCCAGAAACATCCAAT CCL2‑R GCTATGAGCAGCAGGCAC CD44‑F TGGAGGACAGAAAGCCAAGT CD44‑R TTCGCAATGAAACAATCAGTAG PLEKHG2‑M/As‑F CCAAAAGTAAGCCTGTCC PLEKHG2‑M‑R -

Profiling Cellular Processes in Adipose Tissue During Weight Loss Using Time Series Gene Expression
G C A T T A C G G C A T genes Article Profiling Cellular Processes in Adipose Tissue during Weight Loss Using Time Series Gene Expression Samar H. K. Tareen 1,* , Michiel E. Adriaens 1,* , Ilja C. W. Arts 1,2, Theo M. de Kok 1,3, Roel G. Vink 4, Nadia J. T. Roumans 4, Marleen A. van Baak 4, Edwin C. M. Mariman 4, Chris T. Evelo 1,5,* and Martina Kutmon 1,5,* 1 Maastricht Centre for Systems Biology (MaCSBio), Maastricht University, 6211ER Maastricht, The Netherlands; [email protected] (I.C.W.A.); [email protected] (T.M.d.K.) 2 Department of Epidemiology, CARIM School for Cardiovascular Diseases, Maastricht University, 6211ER Maastricht, The Netherlands 3 Department of Toxicogenomics, GROW School of Oncology and Developmental Biology, Maastricht University, 6211ER Maastricht, The Netherlands 4 Department of Human Biology, NUTRIM Research School, Maastricht University, 6211ER Maastricht, The Netherlands; [email protected] (R.G.V.); [email protected] (N.J.T.R.); [email protected] (M.A.v.B.); [email protected] (E.C.M.M.) 5 Department of Bioinformatics—BiGCaT, NUTRIM Research School, Maastricht University, 6211ER Maastricht, The Netherlands * Correspondence: [email protected] (S.H.K.T.); [email protected] (M.E.A.); [email protected] (C.T.E.); [email protected] (M.K.) Received: 28 September 2018; Accepted: 22 October 2018; Published: 29 October 2018 Abstract: Obesity is a global epidemic identified as a major risk factor for multiple chronic diseases and, consequently, diet-induced weight loss is used to counter obesity. -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7, -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Suppl Fig 2 PB2 EUCOMM V3
Maier et al. Supplementary material Semaphorin 4C and 4G are Plexin-B2 ligands required in cerebellar development 2 Supplementary tables 4 Supplementary figures 1 Supplementary Table 1. Exencephaly and postnatal survival of Sema4C mutants (on C57BL/6 background) Genotype Expected frequency Observed frequency Exencephaly Embryonic stages (n=47)* Wild type 25% 25% (12/47) 0% Sema4c+/- 50% 45% (21/47) 0% Sema4c-/- 25% 30% (14/47) 36% (5/14) Postnatal animals (n=417)* Wild type 25% 27% (113/417) Sema4c+/- 50% 61% (253/417) Sema4c-/- 25% 12% (51/417) * Offspring from Sema4c+/- x +/- matings. Embryonic stages scored at E15-E18, postnatal animals scored at P21. 2 Supplementary Table 2. Cerebellar phenotypes of Sema4C and Sema4G mutants (on CD-1 outbred background) Ectopic Fusion of Gap in Normal Gap in IGL granule cells lobules IGL of cerebellum of lobule II in molecular VIII/IX* lobule X Genotype n layer weak strong Wild type 17 100% 0% 0% 0% 0% 0% Sema4c+/- 11 70% 10% 0% 10% 0% 10% Sema4c-/- 10 20% 70% 0% 40% 0% 40% Sema4c+/-; Sema4g+/- 16 55% 18% 0% 18% 0% 18% Sema4c+/-; Sema4g-/- 13 40% 20% 0% 20% 0% 40% Sema4c-/-; Sema4g+/- 10 20% 30% 0% 50% 0% 60% Sema4c-/-; Sema4g-/- 11 0% 55% 0% 55% 55% 82% *Fusions of lobules VIII and IX were scored as “weak” when ectopic granule cells formed a band of cells at the fusion line, and as “strong” when a continuous bridge of granule cells connected the IGL of the two lobules. 3 Supplementary figure legends Maier et al. Suppl. Fig. -

The Human Gene Connectome As a Map of Short Cuts for Morbid Allele Discovery
The human gene connectome as a map of short cuts for morbid allele discovery Yuval Itana,1, Shen-Ying Zhanga,b, Guillaume Vogta,b, Avinash Abhyankara, Melina Hermana, Patrick Nitschkec, Dror Friedd, Lluis Quintana-Murcie, Laurent Abela,b, and Jean-Laurent Casanovaa,b,f aSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; bLaboratory of Human Genetics of Infectious Diseases, Necker Branch, Paris Descartes University, Institut National de la Santé et de la Recherche Médicale U980, Necker Medical School, 75015 Paris, France; cPlateforme Bioinformatique, Université Paris Descartes, 75116 Paris, France; dDepartment of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; eUnit of Human Evolutionary Genetics, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3012, Institut Pasteur, F-75015 Paris, France; and fPediatric Immunology-Hematology Unit, Necker Hospital for Sick Children, 75015 Paris, France Edited* by Bruce Beutler, University of Texas Southwestern Medical Center, Dallas, TX, and approved February 15, 2013 (received for review October 19, 2012) High-throughput genomic data reveal thousands of gene variants to detect a single mutated gene, with the other polymorphic genes per patient, and it is often difficult to determine which of these being of less interest. This goes some way to explaining why, variants underlies disease in a given individual. However, at the despite the abundance of NGS data, the discovery of disease- population level, there may be some degree of phenotypic homo- causing alleles from such data remains somewhat limited. geneity, with alterations of specific physiological pathways under- We developed the human gene connectome (HGC) to over- come this problem. -

A Robust Estimation of Exon Expression To
Risueño et al. BMC Genomics 2014, 15:879 http://www.biomedcentral.com/1471-2164/15/879 METHODOLOGY ARTICLE Open Access A robust estimation of exon expression to identify alternative spliced genes applied to human tissues and cancer samples Alberto Risueño1,5, Beatriz Roson-Burgo1,2, Anna Dolnik3, Jesus M Hernandez-Rivas2,4, Lars Bullinger3 and Javier De Las Rivas1,2* Abstract Background: Accurate analysis of whole-gene expression and individual-exon expression is essential to characterize different transcript isoforms and identify alternative splicing events in human genes. One of the omic technologies widely used in many studies on human samples are the exon-specific expression microarray platforms. Results: Since there are not many validated comparative analyses to identify specific splicing events using data derived from these types of platforms, we have developed an algorithm (called ESLiM) to detect significant changes in exon use, and applied it to a reference dataset of 270 human genes that show alternative expression in different tissues. We compared the results with three other methodological approaches and provided the R source code to be applied elsewhere. The genes positively detected by these analyses also provide a verified subset of human genes that present tissue-regulated isoforms. Furthermore, we performed a validation analysis on human patient samples comparing two different subtypes of acute myeloid leukemia (AML) and we experimentally validated the splicing in several selected genes that showed exons with highly significant signal change. Conclusions: The comparative analyses with other methods using a fair set of human genes that show alternative splicing and the validation on clinical samples demonstrate that the proposed novel algorithm is a reliable tool for detecting differential splicing in exon-level expression data. -

Downloaded, Each with Over 20 Samples for AD-Specific Pathways, Biological Processes, and Driver Each Specific Brain Region in Each Condition
Xiang et al. BMC Medical Genomics 2018, 11(Suppl 6):115 https://doi.org/10.1186/s12920-018-0431-1 RESEARCH Open Access Condition-specific gene co-expression network mining identifies key pathways and regulators in the brain tissue of Alzheimer’s disease patients Shunian Xiang1,2, Zhi Huang4, Wang Tianfu1, Zhi Han3, Christina Y. Yu3,5, Dong Ni1*, Kun Huang3* and Jie Zhang2* From 29th International Conference on Genome Informatics Yunnan, China. 3-5 December 2018 Abstract Background: Gene co-expression network (GCN) mining is a systematic approach to efficiently identify novel disease pathways, predict novel gene functions and search for potential disease biomarkers. However, few studies have systematically identified GCNs in multiple brain transcriptomic data of Alzheimer’s disease (AD) patients and looked for their specific functions. Methods: In this study, we first mined GCN modules from AD and normal brain samples in multiple datasets respectively; then identified gene modules that are specific to AD or normal samples; lastly, condition-specific modules with similar functional enrichments were merged and enriched differentially expressed upstream transcription factors were further examined for the AD/normal-specific modules. Results: We obtained 30 AD-specific modules which showed gain of correlation in AD samples and 31 normal- specific modules with loss of correlation in AD samples compared to normal ones, using the network mining tool lmQCM. Functional and pathway enrichment analysis not only confirmed known gene functional categories related to AD, but also identified novel regulatory factors and pathways. Remarkably, pathway analysis suggested that a variety of viral, bacteria, and parasitic infection pathways are activated in AD samples.